Um alle Funktionen dieser Seite zu nutzen, aktivieren Sie bitte die Cookies in Ihrem Browser.
Meine Merkliste
my.chemie.de
my.chemie.de
Mit einem my.chemie.de-Account haben Sie immer alles im Überblick - und können sich Ihre eigene Website und Ihren individuellen Newsletter konfigurieren.
- Meine Merkliste
- Meine gespeicherte Suche
- Meine gespeicherten Themen
- Meine Newsletter
ChromatografieChromatografie bzw. Chromatographie (griechisch, zu deutsch Farbenschreiben) wird in der Chemie ein Verfahren genannt, das die Auftrennung eines Stoffgemisches durch unterschiedliche Verteilung seiner Einzelbestandteile zwischen einer stationären und einer mobilen Phase erlaubt. Dieses Prinzip wurde erstmals 1901 von dem russischen Botaniker Michail S. Tswett beschrieben, 1903 wurde es zum ersten Mal öffentlich gedruckt beschrieben, 1906 benutzte er erstmals den Begriff „Chromatografie“. Er untersuchte gefärbte pflanzliche Extrakte, zum Beispiel aus Blattmaterial, und konnte daraus durch Chromatografie verschiedene Farbstoffe isolieren. Praktische Anwendung findet diese Methode zum einen in der Produktion zur Isolierung bzw. Reinigung von Substanzen (= präparative Chromatografie), zum anderen in der chemischen Analytik, um Stoffgemische in möglichst einheitliche Inhaltsstoffe zwecks Identifizierung oder mengenmäßiger Bestimmung aufzutrennen. Die Chromatografie ist aus der heutigen organischen Chemie, der Biochemie, der Biotechnologie, der Mikrobiologie, der Lebensmittelchemie, der Umweltchemie und auch der anorganischen Chemie nicht mehr wegzudenken. Weiteres empfehlenswertes Fachwissen
Beschreibung des ChromatografieprozessesDie Chromatografie lässt sich am einfachsten durch einen Vergleich erklären: Ein reißender Fluss kann einiges an Treibgut mit sich führen. Die Geschwindigkeit, mit der das Treibgut weiterbewegt wird, hängt ab
In der Chromatografie werden Substanzgemische (= Treibgut) in der sogenannten Mobilen Phase (= Wasser) auf einer Stationären Phase (= Flussbett) weiterbefördert. Aufgrund der Wechselwirkungen (siehe die Einteilung unter Trennprinzipien) zwischen der Probe, der stationären Phase und der mobilen Phase werden die einzelnen Komponenten unterschiedlich schnell weitertransportiert und können somit voneinander getrennt werden: Ein Gemisch aus Sand, sehr kleinen und etwas größeren Kieselsteinen wird an einer Stelle des Flusses eingebracht; nach beispielsweise hundert Metern kommt zuerst der gesamte Sand an (verteilt auf ein paar Meter) und nach einer gewissen Wartezeit kommen alle kleineren Kieselsteine und noch viel später die größeren, jeweils auf eine gewisse Strecke auseinandergezogen. Dieser bildhafte Vergleich ist sinnvoll für einen ersten Einstieg. Tatsächlich erinnert der Prozess (bei der Chromatografie) eher an einen „digitalen“ Prozess (stop and go). Die Probemoleküle werden entweder mit der mobilen Phase mitgenommen (mit der Geschwindigkeit der mobilen Phase - Analogie wäre ein Floß, das passiv in einem Strom mitgeführt wird) oder sie haften an der stationären Phase (Geschwindigkeit gleich Null). Zwischen diesen beiden Möglichkeiten wechseln sie sehr rasch hin und her (aufgrund der Wärmebewegung erhalten sie ständig Stöße). Der Vergleich mit dem Flussbett könnte auch zu einem weiteren Missverständnis führen: die Verzögerungen, die die verschiedenen Probemoleküle auf dem Weg durch das chromatografische System erleiden, haben nichts mit Reibungsphänomenen zu tun. Basis für das Verständnis sind Unterschiede in der Verteilung (der verschiedenen Molekülsorten A, B, C usw.). Sie entsprechen Unterschieden im Zeitanteil (den die einzelnen Moleküle vom Typ A, B, C usw. im Mittel in der mobilen Phase verbringen). Die Chromatografie schafft es, diese Unterschiede in Geschwindigkeitsunterschiede zu verwandeln und damit für eine Trennung gut nutzbar zu machen. Das könnte man auch als den „Trick“ oder als das Prinzip der Chromatografie bezeichnen. Ansonsten wären diese oft recht kleinen Unterschiede kaum zu nutzen, weder für Trenn- und Reinigungsprozesse noch für Analysen. Anhand eines Beispiels ist es leichter zu verstehen: Wenn sich 45 % der A-Moleküle in der mobilen Phase aufhalten (im Mittel), kommt es auf Grund des dynamischen Gleichgewichtes dazu, dass die individuellen A-Moleküle auch 45 % der Zeit in der mobilen Phase verbringen (im Mittel). Daher wird ihre Geschwindigkeit 45 % der Geschwindigkeit der mobilen Phase betragen (im Mittel). Für gute Ergebnisse bei der Chromatografie ist es entscheidend, dass der Stoffaustausch zwischen den beiden Phasen sehr rasch erfolgt, das heißt die einzelnen Probemoleküle sollen sehr oft zwischen den beiden Phasen hin und her wechseln (Diffusionsprozesse, Wärmebewegung). Eine Voraussetzung dafür ist, dass die Wege, die die Moleküle von der stationären Phase zur mobilen Phase zurückzulegen haben, sehr kurz sind. Falls die stationäre Phase ein Pulver enthält, sollte die Korngröße dieses Pulvers sehr klein sein (zum Beispiel nur wenige Mikrometer). Aus bestimmten Gründen sollten die Pulverkörner auch möglichst einheitlich geformt sein und eine möglichst einheitliche Größe aufweisen (enge Korngrößenverteilung). Prozess
Für die Chromatografie sind die Herstellung des Flusses der mobilen Phase, die Injektion der zu trennenden Probe, die eigentliche Trennung und die Detektion nötig. Das Fließen der mobilen Phase wird entweder mittels Druck (hydraulischen Pumpe, Gasdruck), Kapillarkraft oder durch Anlegen einer elektrischen Spannung erreicht. Die Injektion (= Einbringen des Substanzgemisches in das chromatographische System) erfolgt entweder, bevor der Fluss der Mobilen Phase hergestellt wird (z. B. Dünnschichtchromatographie) oder während die Mobile Phase bereits fließt. Bei einer großen Anzahl von Proben werden bei automatisierbaren Chromatografiearten sogenannte Autosampler (zusammen mit eigenen Datenerfassungssystemen) eingesetzt, die vollautomatisch die Proben injizieren. Anschließend erfolgt die eigentliche Auftrennung des Substanzgemisches auf der Trennstrecke. Ohne Detektion (= Sichtbar machen, wann eine Substanz einen bestimmten Teil des Chromatografiesystems passiert oder wo eine Substanz nach dem Beenden des Prozesses zum Liegen kommt) ist eine Chromatografie nicht denkbar. Für jede Chromatografieart werden verschiedene Detektionsysteme eingesetzt, indem entweder physikalische Eigenschaften (Absorption von Licht, Fluoreszenz, Lichtstreuung, Wärmeleitfähigkeit ..) der Substanzen ausgenutzt werden oder durch chemische Reaktionen ein Signal erhalten wird. Mittels chemischer Reaktionen wird z.B. eine Färbung bei der planaren Chromatografie erreicht (z.B. Aminosäuren mittels Ninhydrin) oder Reaktionen vor dem Auftrennen (Vorsäulenderivatisierung) oder nach dem Auftrennen (Nachsäulenderivatisierung) bei der Säulenchromatographie durchgeführt. Bei der präparativen Chromatografie wird anschließend noch ein Fraktionensammler zum Auffangen der aufgetrennten Substanz benötigt. Bauartbedingt handelt es sich bei chromatographischen Aufreinigungsverfahren immer um Batch-Verfahren. Dies bedeutet, dass immer nur eine bestimmte Menge an Substanz aufgetragen und getrennt werden kann, bevor mit der nächsten Menge fortgefahren werden kann. Dies ist insbesondere bei der Aufarbeitung grosser Mengen problematisch, so dass einige Verfahren entwickelt wurden, um Chromatographie kontinuierlich betreiben zu können: Annulare Chromatographie, TMB (True Moving Bed) Chromatographie und SMB (Simulated Moving Bed) Chromatographie. Definition einiger BegriffeStationäre PhasePhase, die mit den einzelnen Substanzen des Substanzgemisches Wechselwirkungen eingeht und sich nicht bewegt. Der Aufenthalt der Analyten bei ihrer Retention wechselt zwischen mobiler und stationärer Phase (random walk), und verursacht die substanzcharakteristische Retentionszeit. Mobile PhasePhase, in die das Substanzgemisch am Beginn des Trennsystems eingebracht wird und die bewegt wird (Phase an einem festen oder flüssigen Stoff). Mobile Phasen unterscheiden sich in ihrer Elutionsfähigkeit („Stärke“ s. u. „Elutrope Reihe“), dies bedingt unterschiedliche Retentionszeiten und oft auch unterschiedliche Selektivitäten. RetentionVerzögerung von einzelnen Substanzen des Substanzgemisches durch Wechselwirkung mit der stationären Phase. RetentionszeitZeit, die die Moleküle eines reinen Stoffes zum Durchwandern der Säule benötigen (von der Injektion bis zur Detektion). Durchflusszeit (Totzeit)Die Durchflusszeit (früher auch "Totzeit") gibt die Zeit an, die die mobile Phase oder eine nicht zurückgehaltene Substanz benötigt, um die Säule zu durchwandern. Eine nicht zurückgehaltene Substanz (Inertsubstanz) befindet sich nur in einer vernachlässigbar geringen Konzentration in der stationären Phase und durchläuft die Säule daher in der selben Zeit wie die mobile Phase. ElutionElution (von lat. eluere „auswaschen“) ist das Herauslösen oder Verdrängen von adsorbierten Stoffen aus festen oder mit Flüssigkeit getränkten Adsorbentien und Ionenaustauschern durch kontinuierliche Zugabe eines Lösungsmittels (Elutionsmittel = mobiler Phase). Die aus der Trennsäule fließende Lösung wird Eluat genannt. Eine besondere Bedeutung hat dieser Prozess in der Festphasenextraktion. EluentMobile Phase, die die Trennstrecke passiert hat. Eluotrope ReiheAnordnung der als mobile Phase üblichen Lösungsmittel nach ihrer Elutionskraft bei einer Referenzsubstanz (i. d. R. Kieselgel oder Aluminiumoxid). Die Anordnung kann aufsteigend oder absteigend gewählt werden. ChromatographieSäule: In der Chromatografie versteht man unter einer Säule eine hohle Röhre von einem Durchmesser von wenigen Mikrometern bis zu mehreren Metern. Diese Röhre ist entweder mit der stationären Phase komplett gefüllt (gepackte Säule) oder innen dünn beschichtet (Kapillarsäule). Reversed Phase-MechanismusBei der Adsorptionschromatografie gibt es zwei Möglichkeiten ein Substanzgemisch zu trennen:
Im ersten Fall werden lipophile Stoffe leicht, polare schwer eluiert, im Umkehrfall polare leicht eluiert („similia similibus solvuntur“). In der HPLC wird oft eine Gradienteneluierung angewandt (reversed phase), wobei die Zusammensetzung des Lösungsmittels langsam geändert wird (z. B. von 80 % auf 20 % Wasseranteil). So treten Alkane sehr spät und Aminosäuren sehr früh aus der Säule aus und man kann diese Fraktionen herausschneiden. Die chromatografischen Trennmethoden lassen sich nach verschiedenen Gesichtspunkten einteilen: Einteilung nach dem TrennprinzipDas grundlegende Prinzip aller chromatografischen Verfahren ist die oft wiederholte Einstellung eines Gleichgewichtes zwischen einer ruhenden Phase und einer bewegten Phase. Das Gleichgewicht kann sich auf Grund verschiedener physikalisch-chemischer Effekte ausbilden.
Einteilung nach den verwendeten PhasenAuf Grund der mobilen Phasen kann man die Chromatografie in drei Gebiete unterteilen, welche sich nach den Trägern der stationären Phasen oder dem Aggregatzustand der stationären Phasen wieder in unterschiedlich viele Gruppen unterteilen lassen.
Kenngrößen der Chromatografie
bB : Basislinienbreite FWHM : "Full Width at Half Maximum" Fwhm n: Peakkapazität; Gibt an, wieviele Peaks innerhalb eines Intervalls zwischen t0 und dem k-Wert eines bestimmten Peaks theoretisch mit einer Auflösung von R=1 voneinander getrennt werden können.
Trennstufenhöhe HDie Trennstufenhöhe einer chromatografischen Säule ist ein Maß für die Trennleistung der Säule. Als Trennstufe kann man sich den gedachten Abschnitt der Trennsäule, auf dem sich das chromatografische Gleichgewicht einmal einstellt, vorstellen. Je mehr solche Gleichgewichtseinstellungen "auf der Säule Platz haben" umso geringer ist die Trennstufenhöhe und umso höher ist die Trennleistung der Säule. Zur Erlangung einer niedrigen Trennstufenhöhe sind unter analytischen Bedingungen folgende Voraussetzungen nötig:
Zur Ermittlung der Trennstufenhöhe in Abhängigkeit von der Fließgeschwindigkeit des Eluenten kann die sog. Van-Deemter-Gleichung für die HPLC herangezogen werden: wobei:
PeaksymmetrieTheoretisch sollte jede Substanz eine Chromatographiesäule als scharf eluierende Linie verlassen. Aus verschiedenen Gründen besitzen chromatografische Peaks jedoch immer eine gewisse Breite. Im Idealfall weisen sie dabei die Form einer Gauß’schen Glockenkurve auf. In der Praxis kommt es aber häufig vor, dass die Peaks von dieser Idealform abweichen und mehr oder weniger asymmetrisch erscheinen. Eine Asymmetrie, bei der der Frontanstieg des Peaks steiler ist als der Peakabfall, bezeichnet man als „Tailing“, während der Effekt, dass der Anstieg weniger steil ist als der Abfall als „Fronting“ bezeichnet wird. Der Tailingfaktor, der ein Maß für die Peaksymmetrie darstellt, wird bestimmt, in dem man vom Peakmaximum das Lot zur Basislinie fällt, und in einer bestimmten Höhe, meist in 10% der Peakhöhe, die Abstände zur Peakfront (a) und zum Peakende (b) ermittelt. Anschließend wird der Quotient der beiden Werte gebildet, wobei unterschiedliche Berechnungsformeln (z.B. nach IUPAC oder nach USP) in Gebrauch sind:
Praktisches ExperimentChromatografie kann man mit handelsüblichen Mitteln zu Hause durchführen. Man benötigt:
Auf den unteren Rand des Kaffeefilters trägt man Farbe auf und stellt das Papier in eine Schale mit Wasser, sodass sich das Papier mit Wasser vollsaugt. Da die Farbe der Buntstifte wasserlöslich ist, transportiert das Wasser nun die Farbe nach oben. Ein z.B. roter Buntstift ist im Grunde nicht rot, sondern besteht aus einem Gemisch unterschiedlicher Farben, die zusammen rot wirken. In dem Experiment gehen die unterschiedlichen Farbenpigmente nun mit dem Papier eine unterschiedlich starke Wechselwirkung ein und werden dadurch vom Wasser mehr oder weniger schnell transportiert. Daher kann man bald mehrere, verschiedenfarbige Flecken erkennen, die Buntstiftfarben werden chromatografisch getrennt. Um die Abhängigkeit der Trennung vom Lösungsmittel zu testen, kann man dieses Experiment mit dem selben Stift noch einmal mit Wodka, Reinigungsbenzin oder Brennspiritus durchführen und die unterschiedlichen Resultate beobachten. (Das sollte man jedoch nur in gut gelüfteten Räumen tun, da die Dämpfe vom Reinigungsbenzin giftig sind.) Siehe auch
Kategorien: Chromatografie | Trennverfahren |
|
| Dieser Artikel basiert auf dem Artikel Chromatografie aus der freien Enzyklopädie Wikipedia und steht unter der GNU-Lizenz für freie Dokumentation. In der Wikipedia ist eine Liste der Autoren verfügbar. |



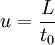
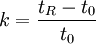
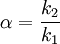
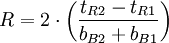 oder
oder
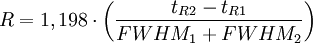
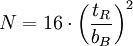 oder
oder
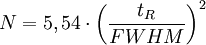
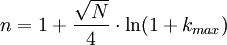
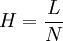
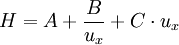
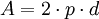 wobei
wobei
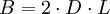 wobei:
wobei: