Um alle Funktionen dieser Seite zu nutzen, aktivieren Sie bitte die Cookies in Ihrem Browser.
Meine Merkliste
my.chemie.de
my.chemie.de
Mit einem my.chemie.de-Account haben Sie immer alles im Überblick - und können sich Ihre eigene Website und Ihren individuellen Newsletter konfigurieren.
- Meine Merkliste
- Meine gespeicherte Suche
- Meine gespeicherten Themen
- Meine Newsletter
Hartree-Fock-MethodeUnter Hartree-Fock-Rechnung versteht man eine Methode der Theoretischen Physik, mit der man hauptsächlich Probleme der Theoretischen Chemie löst. Sie ermöglicht es, Eigenschaften von Vielteilchensystemen, üblicherweise von Atomen oder Molekülen, die zu analytisch nicht mehr lösbaren Gleichungen führen, näherungsweise zu berechnen. Namensgeber hierfür sind Douglas Rayner Hartree und Wladimir Alexandrowitsch Fock. Die Hartree-Fock-Methode ist eine so genannte Ab-initio-Methode, was bedeutet, dass sie ohne empirische Werte auskommt und nur Naturkonstanten benötigt. Produkt-Highlight
FunktionsweiseBei der Aufstellung der Hartree-Fock-Gleichung wird die Wellenfunktion näherungsweise als antisymmetrisiertes Produkt (Slater-Determinante) von Einelektronen-Wellenfunktionen (den Orbitalen, genauer gesagt den Spinorbitalen) angesetzt und darauf das Rayleigh-Ritz-Prinzip angewendet, welches besagt, dass die Energie, die mit einer beliebigen Wellenfunktion eines Systems als Erwartungswert über den Hamiltonoperator dieses Systems berechnet werden kann, immer über der Grundzustandsenergie dieses Systems liegt. Folglich werden die Orbitale so variiert, dass die Energie minimal wird. Es ist aber zu beachten, dass mit der Verringerung der berechneten Energie nicht notwendigerweise auch eine entsprechende qualitative Verbesserung der Wellenfunktion verbunden ist. Bei manchen (open-shell) Molekülen wird statt einer einzigen Slater-Determinante eine Linearkombination mehrerer Slater-Determinanten angesetzt, deren Koeffizienten aber durch die (Spin-)Symmetrie des Systems festgelegt sind. Selbstkonsistente FelderDie Anwendung des Näherungsansatzes für die Wellenfunktion und dem Variationsprinzip resultiert in der Hartree-Fock-Gleichung, welche eine Einelektronen-Differentialgleichung für jedes Orbital darstellt. Diese Gleichung muss iterativ gelöst werden, da die anderen Orbitale als gemitteltes Potenzial auftreten. Man geht dabei so vor, dass man – bildlich gesprochen – auf ein Elektron das gemittelte Potenzial aller anderen Elektronen wirken lässt. So erhält man eine verbesserte Wellenfunktion für dieses Elektron. Dieses Verfahren wird nun für jedes Elektron durchgeführt und so oft wiederholt, bis sich die Wellenfunktionen nicht mehr ändern. Die Wellenfunktionen sind nun selbstkonsistent, stehen also im Einklang mit dem von ihnen erzeugten Feld. Daher wird diese Methode auch als Methode der selbstkonsistenten Felder bezeichnet. Physikalisch gesehen sind Elektronen ununterscheidbar, d. h. kein Elektron ist einem bestimmten Orbital zugeordnet. Dies wird mathematisch durch den Ansatz der Wellenfunktion als Slaterdeterminante (anstatt eines einfachen Hartree-Produkts) gewährleistet. BasissätzeEine direkte numerische Lösung der Hartree-Fock-Gleichung als Differentialgleichung ist bei Atomen und linearen Molekülen möglich. In der Regel werden die Orbitale aber analytisch als Linearkombinationen von Basisfunktionen angesetzt, was wiederum eine Näherung darstellt, die umso besser wird, je größer und intelligenter der Basissatz gewählt wird. Die Basisfunktionen selbst werden wiederum als Produkte von Kugelfunktionen (s, p, d, f Orbitalformen) mit festen Linearkombinationen von Exponentialfunktionen Damit wird die Lösung der Differentialgleichung reduziert auf die (analytische) Berechnung von Integralen über diese Basisfunktionen (die Form ist so gewählt, damit genau das mit vertretbarem Aufwand geht) und die iterative Lösung von Matrixgleichungen (verallgemeinertes Eigenwertproblem, Roothan-Hall-Gleichung) mit den Koeffizienten der Basisfunktionen als zu bestimmende Parameter (für die sehr effiziente Computeralgorithmen existieren). NachteileLeider erreicht die so errechnete Energie nie den exakten Wert, selbst wenn ein beliebig großer Basissatz verwendet werden würde. Bei diesem (theoretischen) Grenzfall wird das sogenannte Hartree-Fock-Limit erreicht. Der Grund dafür ist, dass durch die Verwendung des gemittelten Potenzials die Elektronenkorrelation, also die genaue (instantane) Wechselwirkung der Elektronen untereinander, nicht erfasst wird. Um diesen Makel zu beseitigen, wurden Methoden entwickelt, die in der Lage sind, zumindest einen Teil der Elektronenkorrelation zu erfassen (siehe Artikel Korrelierte Rechnungen). VorteileDie Hartree-Fock-Methode erlaubt bei sehr vielen Molekülen eine gute Bestimmung ihrer "groben" elektronischen Struktur. Sie liefert im Regelfall elektronische Gesamtenergien, die bis auf 0.5% mit den korrekten elektronischen Energien übereinstimmen (zur Berechnung von Energiedifferenzen, wie z.B. Reaktionsenergien, ist sie aber nicht brauchbar), Dipolmomente die auf 20% mit den wirklichen Dipolmomenten übereinstimmen und sehr genaue Verteilungen der Elektronendichte im Molekül. Aufgrund dieser Eigenschaften werden Hartree-Fock-Rechnungen häufig als Ausgangspunkt für genauere Rechnungen verwendet.
Literatur
Kategorien: Quantenphysik | Computerchemie | Theoretische Chemie |
|
| Dieser Artikel basiert auf dem Artikel Hartree-Fock-Methode aus der freien Enzyklopädie Wikipedia und steht unter der GNU-Lizenz für freie Dokumentation. In der Wikipedia ist eine Liste der Autoren verfügbar. |



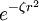 (mit festem
(mit festem 

