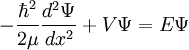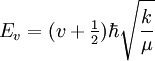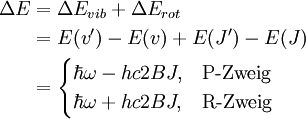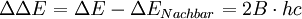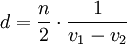Um alle Funktionen dieser Seite zu nutzen, aktivieren Sie bitte die Cookies in Ihrem Browser.
Meine Merkliste
my.chemie.de
my.chemie.de
Mit einem my.chemie.de-Account haben Sie immer alles im Überblick - und können sich Ihre eigene Website und Ihren individuellen Newsletter konfigurieren.
- Meine Merkliste
- Meine gespeicherte Suche
- Meine gespeicherten Themen
- Meine Newsletter
IR-SpektroskopieIR-Spektroskopie (genau: Infrarotspektroskopie) ist ein physikalisches Analyseverfahren, das mit infrarotem Licht (800–500.000 nm) arbeitet. Die IR-Spektroskopie wird zur quantitativen Bestimmung von bekannten Substanzen, deren Identifikation anhand eines Referenzspektrums oder zur Strukturaufklärung unbekannter Substanzen genutzt.
Aus spektroskopischer Sicht wird zwischen dem nahen Infrarot (NIR: 800–2500 nm), dem mittleren oder klassischen (normalen) Infrarot (MIR: 2500–50.000 nm) und dem fernen Infrarot (FIR: 50.000–500.000 nm) unterschieden, da unterschiedliche Phänomene die Absorption im jeweiligen Bereich verursachen. Im FIR-Bereich absorbieren Molekülrotationen und im MIR-Bereich Molekülbindungen. Im NIR-Bereich sind nur noch Obertöne beziehungsweise Kombinationsschwingungen des MIR detektierbar (insbesondere von CH-, OH- und NH-Bindungen). Die IR-Spektroskopie im mittleren Infrarot ist eine der leistungsfähigsten analytischen Techniken in der chemischen Analytik organischer Substanzen. Die Nahinfrarot-Spektroskopie (NIRS) wird trotz ihrer dazu bescheidenen Möglichkeiten zur schnellen Analyse von Stoffgemischen genutzt. In IR-Spektren wird, im Gegensatz zu UV-Spektren, nicht die Absorption sondern die Transmission als Maß für die Durchlässigkeit der Anregungsstrahlung verwendet. Die Transmission wird nach oben zunehmend der vertikalen Achse aufgetragen – Bereiche geringer Durchlässigkeit der IR-Strahlung ergeben einen Ausschlag nach unten.
Weiteres empfehlenswertes Fachwissen
MessprinzipBei der Bestrahlung eines Stoffes mit elektromagnetischen Wellen werden bestimmte Frequenzbereiche absorbiert. Infrarotstrahlung liegt energetisch im Bereich der Schwingungsniveaus von Molekülbindungen, d. h., die Absorption führt zu einer Schwingungsanregung der Bindungen. Sie sind in Form von Ausschlägen im gemessenen Spektrum (Diagramm) sichtbar. Da die dazu notwendigen Energien bzw. Frequenzen charakteristisch für die jeweiligen Bindungen sind, können so auch Materialien identifiziert werden. Die IR-Spektroskopie ist somit strukturklärend. Der einfachste Fall ist ein zweiatomiges Molekül. Bei mehratomigen Molekülen kommt es zur Überlagerung von Grundschwingungen. Dementsprechend sieht man eine Reihe von Absorptionsbanden, die interpretiert werden müssen. Mechanistisches ModellDas denkbar einfachste Modell, welches zur Erklärung von Vibrations- und Rotationsanregungen verwendet werden kann, ist das klassische Modell der Wechselwirkung eines permanenten elektrischen Dipols im elektromagnetischen Feld; Später können in besseren Modellen, wie dem quantenmechanischen Modell, auch Moleküle ohne ein permanentes Dipolmoment beschrieben werden. Zwischen den Atomen und ihren Nachbarn bestehen anziehende und abstoßende Wechselwirkungen. Daher befindet sich der optimale Bindungsabstand im Molekül im Minimum der Potentialfunktion. Mechanistisch lässt sich dies so vorstellen, als wären die Atome durch Federn miteinander verbunden. Die Kraft, die man zur Auslenkung einer Feder braucht, wird mit dem Hookeschen Federgesetz beschrieben. Bringt man nun ein solches Molekül in ein elektrisches Feld, wie es beispielsweise in einem Plattenkondensator besteht, wird sich das Molekül erstens mit seinem Dipolmoment entlang des elektrischen Feldes ausrichten und zweitens seinen Bindungsabstand vergrößern. Wird jetzt Wechselspannung angelegt oder regt man das Molekül mit einer elektromagnetischen Welle an, fangen die an den Bindungen „hängenden“ funktionellen Gruppen zu schwingen und zu rotieren an. Das mechanistische Modell ist allerdings nur sehr begrenzt tauglich, da es beispielsweise nicht erklärt, warum nur diskrete Energien zur IR-Anregung zugelassen sind und warum auch Moleküle ohne permanentes Dipolmoment eine IR-Absorption zeigen. Quantenmechanisches Modell
Wie auch im mechanistischen Modell ist die Grundlage des quantenmechanischen Modells der Vibrations- und Rotationsanregung die Potentialfunktion. Die Potentialfunktion lässt sich in ihrem Minimum gut durch eine Parabel annähern. Eine solche Parabel ergibt sich aus der Integration des Hookschen Federgesetzes. Wird jetzt das Molekül mit elektromagnetischer Strahlung angeregt, kann das Molekül anfangen zu schwingen, wenn die Energie ausreicht, es aus seinem Grundzustand in den ersten angeregten Schwingungszustand zu heben. Um diese Energie zu bestimmen, muss die Schrödinger-Gleichung für dieses Potential gelöst werden. Nach Abtrennung der Relativbewegung von Atomkernen und Elektronen ergibt sich als Lösung der Schrödinger-Gleichung ein Zusammenhang zwischen der benötigten Energie, Bindungsstärke (k) und der reduzierten Masse (μ). Anders als beim klassischen harmonischen Oszillator ist im Quantenmechanischen Fall die Schwingungsenergie durch die Schwingungsquantenzahl v gequantelt. Schrödinger-Gleichung:
Lösung der Schrödinger-Gleichung:
Das ideale Rotations-Schwingungs-Spektrum am Beispiel von Chlorwasserstoff
Chlorwasserstoff (HCl) ist ein zweiatomiges Molekül mit einem ausgeprägten Dipolmoment. Das HCl-Molekül kann vereinfacht als ein linearer Kreisel angesehen werden. Die Quantenmechanik liefert für ein lineares Molekül die spezielle Auswahlregel für einen Rotationsübergang:
wobei J die Rotationsquantenzahl darstellt. Wenn das Dipolmoment parallel zur Hauptrotationsachse liegt (wie z. B. beim Ammoniak), wäre auch ΔJ=0 möglich. Für einen Schwingungsübergang des harmonischen Oszillators lautet die spezielle Auswahlregel:
Die Übergänge zwischen den verschiedenen Rotationsniveaus (J = 0, 1, 2, …) des Schwingungsgrundzustandes (v = 0) und den Rotationsniveaus (J' = 0, 1, 2, …) des angeregten Schwingungszustandes (v' = 1) können in zwei Gruppen, den R-Zweig (rechts) für Übergange mit ΔJ = +1 und den P-Zweig (links) für Übergänge mit ΔJ = -1, unterteilt werden und sind in der Abbildung skizziert. Da ΔJ = 0 für HCl nicht erlaubt ist, erscheint auch kein Q-Zweig. Im oberen Bild ist das Energieniveauschema abgebildet. Anhand der Pfeillänge ist deutlich zu erkennen, dass jeder Übergang eine Energiezufuhr von erfordert. Für die Energiedifferenz zum benachbarten Übergang gilt damit Im idealen Rotationsschwingungs-spektrum (unteres Bild) haben daher die Peaks einen konstanten Abstand von 2B. Im Experiment weisen die Peaks etwas andere Abstände auf, da das Modell nicht:
Das reale Rotations-Schwingungs-Spektrum am Beispiel von Chlorwasserstoff
Das reale Rotations-Schwingungs-Spektrum weicht von dem idealen ab aufgrund der Einschränkungen der Messmethoden und aufgrund von Kopplungen. Die Abstände der Peaks (Amplitudenausschlag im Diagramm) betragen nicht wie im idealen Fall genau 2B, sondern werden mit steigender Wellenzahl kleiner, sowohl im P-Zweig als auch im R-Zweig. Das nebenstehende Diagramm zeigt ein aufgenommenes Spektrum von Chlorwasserstoff. Die Feinstruktur des Spektrums ist gut für beide Isotope des Chlors zu erkennen. MolekülschwingungenIn anorganischen und organischen Substanzen treten bei Absorption von Strahlung aus dem infraroten Bereich mechanische Schwingungen auf. Es können drei verschiedene Arten von Schwingungen unterschieden werden:
Da Molekülschwingungen bestimmter Atomgruppen im Bereich von 4000–1500 cm-1 besonders charakteristisch sind, eignet sich die IR-Spektroskopie zur Bestimmung der funktionellen Gruppen des untersuchten Moleküls. Zusätzlich ist das gesamte Spektrum und besonders der Fingerprintbereich, im Bereich von 1500–600 cm-1, zwar nicht für buchstäblich jede Substanz charakteristisch, wie oft behauptet, bietet aber oft wertvolle Anhaltspunkte zur Identifikation. Enantiomere chiraler Verbindungen zeigen nicht grundsätzlich unterschiedliche Banden im Fingerprintbereich und keinesfalls in irgendeinem anderen. Absorption von IR-StrahlungWechselwirkung zwischen elektromagnetischer Strahlung und dem Molekül kann nur dann auftreten, wenn im Molekül bewegte elektrische Ladung zur Verfügung steht. Das ist immer dann der Fall, wenn das Molekül entweder ein veränderbares oder ein induzierbares Dipolmoment aufweist (IR-aktiv). In Molekülen mit Schwingungen symmetrisch zum Symmetriezentrum treten keine Änderungen des Dipolmoments auf (IR-inaktiv). Lage der IR-AbsorptionsbandenStärkere chemische Bindungen und Atome kleinerer Masse verursachen im IR-Spektrum Absorptionsmaxima bei großen Wellenzahlen (hohe Energie), große Massen hingegen verursachen IR Absorptionsmaxima bei kleinen Wellenzahlen (niedrige Energie) (siehe Deuterierung). Die Energie ist proportional zum Quadrat des permanenten Dipolmomentes. Daher liefern polare Moleküle intensive Rotationsübergänge. Vergleicht man allerdings die Amplituden der Peaks eines einzelnen Moleküls miteinander, fällt auf, dass die Stärke der Übergänge mit wachsenden J zunächst schnell zunehmen, durch ein Maximum gehen und schließlich für große J wieder abfallen. Die Ursache dafür ist, dass die Stärke die Entartungen der verschiedenen Rotationszustände und die Besetzungszahlen der Rotationsniveaus im Ausgangszustand widerspiegelt. Der Grad der Entartung nimmt mit steigendem J zu, was zu einer höheren Energie führt. Andererseits nehmen die Besetzungszahlen mit steigender Energie ab, was schließlich zu einer Abnahme der Strahlungsstärke führt. IR-Spektren werden dahingehend interpretiert, dass man aus der Kurve des gemessenen IR-Spektrums die Molekülgestalt herauszufinden versucht. Dabei misst man die verschiedenen Schwingungsvarianten der Moleküle. Ein typisches IR-Spektrum reicht von 4000 cm-1 bis 400 cm-1 (Wellenzahl). Die Wellenzahl wird in der etwas ungewöhnlichen Einheit cm-1 angegeben. Man kann aus der Wellenzahl allerdings bequem die Anregungsenergie berechnen, indem man die Wellenzahl mit h und c (Plancksches Wirkungsquantum und Lichtgeschwindigkeit) mulitpliziert. Bei einem IR-Spektrum hinterlässt jedes Molekül ein typisches Muster. Ab etwa 1500 cm-1 abwärts lassen sich die Ursachen für das Spektrum kaum mehr auf bestimmte Molekülgruppen zurückführen. Dies ist der sogenannte Fingerprint-Bereich. Mit ihm lässt sich die Struktur eines Moleküls zwar nicht bestimmen, allerdings ist dieser Bereich eben eine Art „Fingerabdruck“ des Moleküls. Da in der Praxis natürlich meist keine Reinsubstanzen, sondern Gemische vorliegen, kann aus einem IR-Spektrum oft keine exakte Zusammensetzung hergeleitet werden. Allerdings genügt es oft schon zu wissen um welche Stoffgruppe es sich handelt. Schwingungsdaten von wichtigen MolekülgruppenDie folgende Auflistung enthält exemplarisch einige Molekülgruppen und die zugehörigen Bereiche der Absorptionsbanden:
IR-Spektroskopie-TechnikenTransmissionDie heute am häufigsten eingesetzte Methode ist die Transmission. Dabei wird eine Probe vorbereitet, etwa indem sie in KBr gepresst und mit IR-Strahlung beschossen wird. Ein Teil dieser Strahlung durchdringt die Probe, ein anderer Teil wird von der Probe absorbiert. Reflexion und Absorption (IRAS)Eine sehr genaue Messmethode ist die Reflexion und Absorption. Hierbei wird IR-Strahlung fast parallel auf die Probe geschickt. Die p-Komponente des Lichtes regt die Probe zur Schwingung an. Bei dieser Messmethode ist die Intensität sehr viel geringer als bei der Transmission. Diffuse ReflexionMit der diffuse Reflexionsspektroskopie (siehe DRIFTS) können Pulver mit IR-Strahlung beleuchtet werden. Die diffus reflektierte Strahlung wird von einer reflektierenden Kugel nochmals reflektiert und im Detektor gebündelt. Die Strahlung wird nun detektiert und integriert. Das entstehende Spektrum wird mit der Kubelka-Munk-Funktion ausgewertet und man erhält schließlich ein Absorptionsspektrum. Abgeschwächte Totalreflexion (ATR)Bei ATR-Spektroskopie wird die Strahlung in einem Lichtwellenleiter in Totalreflexion geführt. Dabei bildet sich an der Oberfläche dieses Lichtwellenleiters ein evaneszentes Feld aus. Proben werden nun sehr nah an die Oberfläche gebracht, wo sie mit dem evaneszenten Feld wechselwirken können. Die Strahlung, die aus dem Wellenleiter austritt, wird mit einem Spektrometer ausgewertet. Es ergeben sich ähnliche Spektren wie bei der Transmissionsspektroskopie. Diese Methode eignet sich für feste und flüssige Proben. Emission (IRES)Hierbei wird eine Probe auf mindestens 100 °C erhitzt und die emitierte Strahlung gemessen. Diese Technik ist noch nicht sehr gut erforscht. SchichtdickenmessungDa es bei planparallelen dünnen Proben, z. B. Folien zu Interferenzen kommt, wird das eigentliche Spektrum von einem sinusförmigen Wellenzug überlagert. Aus der Lage der Maxima kann man die Dicke der Folie berechnen. mit n = Anzahl der Maxima zwischen v1 und v2 Eliminieren kann man die Interferenzen bei Folien indem man sie knittert oder aufrauht. Literatur
Siehe auch
Kategorien: Physikalisches Analyseverfahren | Spektroskopie |
|||||
| Dieser Artikel basiert auf dem Artikel IR-Spektroskopie aus der freien Enzyklopädie Wikipedia und steht unter der GNU-Lizenz für freie Dokumentation. In der Wikipedia ist eine Liste der Autoren verfügbar. |