Um alle Funktionen dieser Seite zu nutzen, aktivieren Sie bitte die Cookies in Ihrem Browser.
Meine Merkliste
my.chemie.de
my.chemie.de
Mit einem my.chemie.de-Account haben Sie immer alles im Überblick - und können sich Ihre eigene Website und Ihren individuellen Newsletter konfigurieren.
- Meine Merkliste
- Meine gespeicherte Suche
- Meine gespeicherten Themen
- Meine Newsletter
ChemoinformatikChemoinformatik, Cheminformatik oder Chemieinformatik (engl. chemoinformatics, cheminformatics, chemical informatics oder chemiinformatics) bezeichnet einen Wissenschaftszweig, der das Gebiet der Chemie mit Methoden der Informatik verbindet mit dem Ziel, Methoden zur Berechnung von Moleküleigenschaften zu entwickeln und anzuwenden. Zu den Urvätern gehören u. a. Paul deMain, Johann Gasteiger, Jure Zupan, Ivar Ugi. Der Begriff „Chemoinformatik“ ist relativ jung, während die älteren Termini Computerchemie (abgel. von Engl. Computational Chemistry) und chemische Graphentheorie das gleiche Gebiet bezeichnen (Lit.: Bonchev/Rouvray, 1990). Computerchemie wird heutzutage eher als ein Teilgebiet der Theoretischen Chemie und der Quantenchemie begriffen. Produkt-Highlight
GrundlagenDie Chemoinformatik beschäftigt sich mit Berechnungen an digitalen Repräsentationen von Molekülstrukturen. Molekülstrukturen können als Graphen aufgefasst werden. Als ihre Repräsentation ist für viele Anwendungen bereits die sog. Bindungstabelle (engl. connection table) ausreichend, in der die Art der Verknüpfungen (Bindungen) zwischen den einzelnen Atomen eines Moleküls abgelegt ist. Erst für weitergehende Betrachtungen kann die Einbeziehung von zweidimensionalen (2D-) bzw. dreidimensionalen (3D-) Koordinaten notwendig werden. Letztere werden insbesondere benötigt, wenn, etwa im Bereich der Medizinischen Chemie, Wechselwirkungen mit Biomolekülen wie Proteinen untersucht werden sollen. Repräsentation chemischer StrukturenDie Repräsentation chemischer Strukturen ist eine der grundlegenden Fragestellungen. Für einen Großteil der Anwendungen hat sich die Darstellung als Bindungstabelle (Connection Table) basierend auf der Valenzstrukturtheorie durchgesetzt. Als Beispiel einer Bindungstabelle sei hier Benzol im Standardformat Molfile der Firma MDL angegeben. Die Zeilen 5-10 enthalten die 2D-Koordinaten und Elementbezeichner der Atome, die Zeilen 11-16 die Bindungstabelle mit den Ausgangs- und Endatomen jeder Bindung sowie dem Bindungstyp. Die Null-Spalten enthalten mögliche weitere Bezeichner.
Benzene
-ISIS- 09270713002D
6 6 0 0 0 0 0 0 0 0999 V2000
-1.9765 0.3000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.9818 -0.5232 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.2726 -0.9375 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.5577 -0.5299 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.5564 0.2965 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.2661 0.7072 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0 0 0 0
3 4 2 0 0 0 0
4 5 1 0 0 0 0
2 3 1 0 0 0 0
5 6 2 0 0 0 0
6 1 1 0 0 0 0
M END
Zusätzlich zur Bindungstabelle können 3D-Koordinaten für real existierende Moleküle über Röntgenstrukturanalyse ermittelt werden. Wo dies nicht möglich ist oder ein Molekül physisch nicht existent ist, können 3D-Koordinaten zumindest näherungsweise auch unmittelbar aus der Bindungstabelle durch iterative Energie-Minimierungsrechnungen für verschiedene Konformationen eines Moleküls erzeugt werden. 2D-Koordinaten dienen in der Regel allein der Veranschaulichung eines Moleküls und müssen daher hauptsächlich ästhetischen Ansprüchen genügen. Sie werden ebenfalls unmittelbar aus der Bindungstabelle nach allgemein anerkannten chemischen Zeichenregeln errechnet geben nur in den seltensten Fällen die tatsächlichen Gegebenheiten in einem Molekül wieder. Die Größe des gesamten theoretischen chemischen Raumes, welcher aus allen denkbaren (virtuellen) Molekülstrukturen besteht, wird auf etwa 1062 Moleküle geschätzt und ist damit weit größer als die Menge der bisher real synthetisierten Moleküle (Lit.: Lahana, 1999). Mit Hilfe von Computer-basierten Methoden lassen sich aber unter Umständen viele Millionen Moleküle bereits theoretisch (in silico) analysieren, ohne diese zunächst für Messungen im Labor synthetisieren zu müssen. MethodenVerfahren, die keine empirischen Parameter benötigen, werden als ab-initio-Methoden bezeichnet. Semiempirische Verfahren enthalten empirische Größen und weitere semiempirische Parameter, die durch theoretische Vorgehensweisen bestimmt wurden, jedoch keinen Bezug zu messbaren Größen mehr haben. Prinzipiell sind ab initio-Verfahren für kleinere Moleküle geeignet. Semiempirische Verfahren spielen ihre Stärke bei mittelgroßen (100 Atome) Molekülen aus. Beispiele für semiempirische Methoden sind MNDO und AM1. ab-initio-MethodenDie Güte, mit denen ab-initio-Verfahren die Eigenschaften von Molekülen berechnen können, hängt im wesentlichen vom Basissatz der Atome ab, d. h. wie gut und mit wievielen einzelnen Funktionen die Atomorbitale dargestellt werden. Die Verbesserung des Basissatzes in ab initio Verfahren konvergiert Richtung Hartree-Fock-Limit, ist aber für alle Mehrelektronensysteme noch deutlich von der Wirklichkeit entfernt. Grund hierfür ist der Effekt der sogenannten Elektronenkorrelation: Die Bewegung der Elektronen im Molekül ist nicht unabhängig voneinander, sondern je näher sich zwei Elektronen kommen, desto stärker stoßen sie sich ab. Ab initio Verfahren, die auch die Elektronenkorrelation berücksichtigen, sind deutlich aufwändiger (derzeit maximal zehn Elektronen im System möglich), liefern jedoch die besten Resultate. Man behilft sich mit einem Kompromiss und bezieht die Elektronenkorrelation näherungsweise ein. Beispiele für solche Verfahren sind: DFT (Dichtefunktionaltheorie), Møller-Plesset-Störungstheorie, CI (Configuration Interaction), CC (Coupled Cluster), MCSCF (Multiconfiguration Self Consistent Field). Semiempirische VerfahrenBei semiempirischen Verfahren wird ein Großteil der Integrale des Hartree-Fock-Formalismus vernachlässigt, andere werden durch spektroskopische Werte, Parameter oder parametrisierte Funktionen angenähert. Grund für diese Approximation war die geringe Rechenkapazität früherer Zeiten. Um die theoretischen Erkenntnisse dennoch auf chemische Fragestellungen anwenden zu können, musste der vorhandene Formalismus vereinfacht werden. Die Hückel-Näherung ist der einfachste semiempirische Ansatz, da sie gar keine Integrale berechnet. Allerdings ist sie auch nur auf π-Elektronensysteme anwendbar. Die Theorie wurde später auch auf σ-Systeme erweitert (Extended Hückel Theory, EHT). Etablierte Methoden, die auch heutzutage noch häufig angewendet werden, gehören zur Klasse der NDDO-Näherung (Neglect of Diatomic Differential Overlap): MNDO (Modified Neglect of Differential Overlap), AM1 (Austin Model 1), PM3 (Parametrised Method 3). Für kritische Berechnungen sind semiempirische Methoden mit CI (Configuration Interaction) und MCSCF (Multiconfiguration Self Consistent Field) kombiniert worden. Mit solchen Verfahren sind dann beispielsweise Reaktionsbarrieren und ganze Energieprofile komplexer Reaktionen berechenbar (MNDO/CI, MNDO/MCSCF). Die Grenzen semiempirischer Methoden liegen in ihrer Parametrisierung: Eigentlich können mit der fertigen Methoden nur Systeme gerechnet werden, die in ähnlicher Weise im Parametrisierungsdatensatz vorhanden waren. Molekular mechanische VerfahrenKraftfeldprogramme verwenden einen klassisch-mechanischen Ansatz: Bindungen zwischen zwei Atomen A und B werden dabei einfach als Sprungfeder angenähert und mit einem harmonischen Potential beschrieben (Hook'sches Gesetz):
Da eine Doppelbindung zwischen zwei Kohlenstoffatomen eine andere Stärke und Gleichgewichtslänge als eine Einfachbindung besitzt, werden unterschiedliche Parametersätze benötigt (Kraftkonstante kAB und Ruhelage Kraftfelder ermöglichen die Geometrieoptimierung sehr großer (Bio)moleküle (z. B. Proteine) und werden hauptsächlich für Moleküldynamik- oder Monte-Carlo-Simulationen verwendet. AnwendungenEs gibt verschiedene wichtige Themen innerhalb des Gebietes. Eine Auswahl:
Im Folgenden werden ausgewählte Anwendungsbeispiele genauer dargestellt. Quantitative Struktur-Wirkungs-BeziehungMit Hilfe geeigneter Algorithmen werden Kodierungen für Moleküle entwickelt. Durch Induktion können neue Hypothesen über molekulare Eigenschaften erstellt werden, wie z. B. die Bioverfügbarkeit oder die Fähigkeit einer Substanz, die Funktion eines bestimmten Proteins im Organismus zu hemmen oder zu verstärken. Siehe auch QSAR. LeitstrukturoptimierungDurch geeignete chemische und biologische Hypothesen lässt sich dieser chemische Raum auf wenige Kandidaten reduzieren, die dann im Labor synthetisiert und klinisch getestet werden. Aus diesem Grund spielt die Cheminformatik im Bereich der pharmazeutischen Chemie und der Medizinalchemie eine große Rolle zur Optimierung von Leitstrukturen. ThermodynamikIn der technischen Chemie werden Gruppenbeitragsmethoden verwendet, um Stoffeigenschaften wie Normalsiedepunkte, kritische Daten, Oberflächenspannungen u. a. m. abzuschätzen. Molecular ModelingDas Molecular Modeling beschäftigt sich z. B. mit der Schaffung von Modellen unbekannter Makromoleküle anhand der Vorlage (Template) ähnlicher, bekannter Moleküle (Homologiemodeling), der Wechselwirkung zwischen kleinen und großen Molekülen (Rezeptordocking), wodurch QSAR möglich wird, der Moleküldynamik sowie die Entwicklung energitisch minimierter 3D-Strukturen von Molekülen (Bergsteigeralgorithmus, Simulierte Abkühlung, Molekülmechanik etc.). Es geht also darum, aufgrund bekannter Strukturen Modelle von unbekannten Strukturen zu entwickeln, um so eine QSAR zu ermöglichen. Verwandte GebieteEs gibt einen starken Bezug zur Analytischen Chemie und zur Chemometrie. Die Struktur-Eigenschafts-Beziehungen (z. B. Spektrenkorrelation) spielen eine zentrale Rolle. Aufgrund vergleichbarer Arbeitsweise existiert eine enge Beziehung zur Computerphysik, wodurch eine klare Trennung häufig nicht eindeutig gegeben ist. SoftwarepaketeDie Programme der Computerchemie basieren auf den verschiedensten quantenchemischen Methoden zur Lösung der molekularen Schrödingergleichung. Grundsätzlich lassen sich zwei Ansätze unterscheiden: Semiempirische Verfahren und ab-initio-Verfahren. Alle beschriebenen Verfahren und Methoden sind in gängigen Softwarepaketen verfügbar. Beispiele hierfür: ACES, GAUSSIAN, GAMESS, MOLPRO, Spartan, TURBOMOLE, Cerius2 und Jaguar. ArgusLab eigent sich als freiverfügbares Programm zum Einstieg in der Computerchemie. Die Herausforderung für den Anwender dieser Software ist es, das am besten geeignete Modell für seine Problemstellung zu finden und die Ergebnisse im Gültigkeitsbereich der Modelle zu interpretieren. Literatur
Siehe auch
|
|
| Dieser Artikel basiert auf dem Artikel Chemoinformatik aus der freien Enzyklopädie Wikipedia und steht unter der GNU-Lizenz für freie Dokumentation. In der Wikipedia ist eine Liste der Autoren verfügbar. |



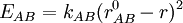
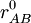 ). Man verwendet daher zur Kennzeichnung der Atome keine einfachen Elemente mehr, sondern Atomtypen.
Ähnliche Ansätze gibt es für Bindungs- und Torsionswinkel. Elektrostatische (
). Man verwendet daher zur Kennzeichnung der Atome keine einfachen Elemente mehr, sondern Atomtypen.
Ähnliche Ansätze gibt es für Bindungs- und Torsionswinkel. Elektrostatische (Zuletzt angesehen


