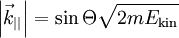Um alle Funktionen dieser Seite zu nutzen, aktivieren Sie bitte die Cookies in Ihrem Browser.
Meine Merkliste
my.chemie.de
my.chemie.de
Mit einem my.chemie.de-Account haben Sie immer alles im Überblick - und können sich Ihre eigene Website und Ihren individuellen Newsletter konfigurieren.
- Meine Merkliste
- Meine gespeicherte Suche
- Meine gespeicherten Themen
- Meine Newsletter
PhotoelektronenspektroskopieDie Photoelektronenspektroskopie (PES) oder Photoemissionsspektroskopie (kurz Photoemission) beruht auf dem äußeren Photoeffekt, bei dem durch elektromagnetische Strahlung Photoelektronen aus dem Festkörper ausgelöst werden. Die Bestimmung der kinetischen Energie dieser Elektronen (Spektroskopie) erlaubt Rückschlüsse auf die chemische Zusammensetzung und elektronische Beschaffenheit des untersuchten Festkörpers. Die PES zählt heute in der Festkörperphysik und in einigen angrenzenden Gebieten wie der Oberflächenphysik und der Werkstoffforschung zu den zentralen Verfahren bei der Untersuchung der Struktur der besetzten elektronischen Zustände. Außerdem haben apparative Weiterentwicklungen während der letzten zehn Jahre neue Untersuchungsfelder in der Grundlagenforschung für die Photoelektronenspektroskopie erschlossen. Traditionell wird die Photoelektronenspektroskopie in UPS (engl. ultraviolet PES), XPS (engl. x-ray PES) und ARPES (engl. angle-resolved PES) unterschieden. Dabei beschreibt UPS die Spektroskopie mit Valenzelektronen, die vorwiegend Aussagen über chemische Verbindungen und elektronische Eigenschaften eines Materials machen kann. XPS wird auch ESCA (engl. electron spectroscopy for chemical analysis) genannt und beschreibt die chemische Analyse eines Materials, beispielsweise in Bezug auf die atomare Zusammensetzung. Mit ARPES wird die elektronische Struktur eines Festkörpers untersucht. Insbesondere eignet sich diese Messmethode zum Vergleich der theoretisch gerechneten, mit dem realen Verlauf der Spektralfunktion des Elektronensystems. Produkt-HighlightGeschichteDer äußere photoelektrische Effekt wurde experimentell 1887 von Heinrich Hertz [1] und Wilhelm Hallwachs [2] entdeckt und später von Albert Einstein [3] erklärt (Physiknobelpreis 1921). Hallwachs erkannte, dass nicht die Intensität des Lichts, sondern dessen Frequenz darüber entscheidet, ob Elektronen aus der Oberfläche einer Photokathode herausgelöst werden können. Einstein führte den Begriff des Lichtquants (Photon) ein und zeigte, dass dessen Energie, die sich – wie Max Planck für die Wärmestrahlung zuvor entdeckte – unmittelbar aus der Lichtfrequenz ν ergibt, mindestens so groß wie die Austrittsarbeit Φ der Festkörperoberfläche sein muss. Seine lichtelektrische Gleichung ergibt die kinetische Energie eines Photoelektrons Ekin, das durch ein Photon der Energie EPhoton aus einem Zustand mit der Bindungsenergie EB angeregt wird. Die Photoelektronenspektroskopie wurde ab 1960 von Kai Siegbahn in Uppsala systematisch zu einer wichtigen experimentellen Untersuchungsmethode der Oberflächen- und Festkörperphysik entwickelt, wofür er auch 1981 den Nobelpreis erhielt. Die zugrunde liegende Idee bestand darin, die Energieverteilung der Elektronen im Festkörper durch Photoemissionsanregung in eine Intensitätsverteilung I(Ekin) von Photoelektronen einer bestimmten Energie Ekin zu überführen. Die kinetische Energie der Photoelektronen lässt sich dann mittels geeigneter magnetischer oder elektrostatischer Analysatoren messen (spektroskopieren). Zur Anregung der Photoelektronen verwendete er zwei verschiedene Typen von Lichtquellen, die auch heute noch im Laborbetrieb üblich sind, die Gasentladungslampe und die Röntgenanode. Die in diesen Quellen entstehende und für die PES genutzte Strahlung liegt im harten Ultraviolett-Bereich bzw. im weichen Röntgenbereich. Entsprechend der Energie der verwendeten Strahlung unterscheidet man bis heute die Photoelektronenspektroskopie in UPS (Ultraviolett-Photoelektronenspektroskopie) und XPS, nach der englischen Bezeichnung x-ray für Röntgenstrahlung. Die Energieauflösung der ersten eingesetzten Instrumente betrug typischerweise zwischen 1 und 2 eV im XPS- und 100 meV oder weniger im UPS-Bereich. Eine wesentliche Entdeckung von Siegbahn war, dass die Spektren der Rumpfelektronen von der chemischen Umgebung des untersuchten Systems abhängen. In den XPS-Spektren desselben Elementes zeigen sich, je nachdem in welcher chemischen Form es vorliegt, Unterschiede in der Bindungsenergie eines Rumpfelektrons von bis zu einigen Elektronenvolt, und in vielen Fällen kann auch die Form der Spektren Aufschluss über den Valenzzustand eines Elementes geben. Auf diesen Beobachtungen und den daraus resultierenden Anwendungsmöglichkeiten gründet sich der zweite Name von XPS, ESCA (Electron Spectroscopy for Chemical Analysis). Die Methode, Moleküle in der Gasphase mittels ultravioletten Lichts zu studieren, wurde von David W. Turner entwickelt und in einer Serie von Veröffentlichungen von 1962 bis 1970 beschrieben. Als Lichtquelle verwendete er eine He-Gasentladungslampe (E = 21,22 eV) deren Emission im ultravioletten Bereich liegt. Mit dieser Quelle erreichte die Gruppe um Turner eine Energieauflösung von ca. 0,02 eV und war damit in der Lage die Energie von Molekülorbitalen sehr genau zu bestimmen und mit theoretischen Werten der damals aktuell entwickelten Quantenchemie zu vergleichen. Aufgrund der Anregung mittels UV-Licht wurde diese Messmethode – in Anlehnung an XPS – UPS genannt. Theoretische BeschreibungDie Photoelektronenspektroskopie ist eine Messmethode, die auf dem äußeren Photoeffekt beruht. Bestrahlt man ein Gas oder einen Festkörper mit Licht der bekannten Energie EPhoton, so werden Elektronen der kinetischen Energie Ekin frei. Einstein konnte mit seiner lichtelektrischen Gleichung den Zusammenhang zwischen der eingestrahlten Photonenenergie und der kinetischen Energie der Elektronen herstellen: Ekin = EPhoton − EB − Φ Über diese Gleichung können bei bekannter Photonenenergie und gemessener Elektronenenergie Aussagen über die Bindungsverhältnisse der Elektronen in dem untersuchten Material gemacht werden. Die Bindungsenergie EB bezieht sich dabei auf das chemische Potenzial des Festkörpers, und die Austrittsarbeit Φ des Probenmaterials ist eine charakteristische, material- bzw. oberflächenspezifische Größe, die sich mittels des äußeren Photoeffekts bestimmen lässt (siehe Bild 2). In einer Näherung nach Koopman wird angenommen, dass sich die Lage der Energieniveaus eines Atoms oder Moleküls bei seiner Ionisierung nicht ändert. Dadurch ist die Ionisationsenergie I für das höchste besetzten Orbital (HOMO: Highest Occupied Molecular Orbital) gleich der negativen Orbitalenergie ε, also der Bindungsenergie. Bei genauer Betrachtung dieser Energie bei Rumpfniveauelektronen kann so auf die Atomsorte geschlossen werden und man erhält aus der quantitativen Analyse die chemische Zusammensetzung (Stöchiometrie) der Probe und bis zu einem gewissen Grad auch die chemischen Bindungsverhältnisse im untersuchten Festkörper. Außerdem erlaubt die Analyse der Bindungsenergie der Valenzband- und Leitungselektronen eine sehr detaillierte Untersuchung des Anregungsspektrums des Elektronensystems kristalliner Festkörper. Die zusätzliche Bestimmung des Winkels, unter dem die Photoelektronen einen Festkörper verlassen, erlaubt eine genauere Untersuchung der Valenzbandstrukturen kristalliner Festkörper, wobei man sich die Impulserhaltung beim Photoemissionsprozess zunutze macht. Aufgrund des Zusammenhangs zwischen dem Impuls des Photoelektrons und dem Wellenvektor eines Bloch-Elektrons ist es dabei möglich, aus der Winkelabhängigkeit der Spektren auf die Dispersionsrelationen der Valenzzustände zu schließen. Diese winkelaufgelöste Photoelektronenspektroskopie wird auch kurz ARPES genannt (angular resolved photoelectron spectroscopy). Bei Metallen beinhalten die elektronischen Dispersionsrelationen die Information über die Form der Fermifläche, die sich auch mit einer Reihe anderer Methoden ermitteln lässt, wie z. B. dem de-Haas-van-Alphen-, Schubnikow-de-Haas- oder dem anomalen Skineffekt. Die genannten Methoden müssen allerdings bei möglichst tiefen Temperaturen an hoch reinen Einkristallen durchgeführt werden, wohingegen die ARPES auch bei Raumtemperatur und vergleichsweise defektreichen Kristallen angewendet werden kann. Messmethoden der PhotoelektronenspektroskopieChemische Analyse (XPS, ESCA)XPS (X-ray photoelectron spectroscopy) ist eine etablierte Methode, um die chemische Zusammensetzung von Festkörpern bzw. deren Oberfläche zerstörungsfrei zu bestimmen. Man erhält dabei zunächst eine Antwort auf die Frage der qualitativen Elementaranalyse, also aus welchen Elementen der Festkörper besteht. Lediglich Wasserstoff und Helium können im Allgemeinen nicht nachgewiesen werden. MessprinzipDie Bindungsenergie EB, die aus der kinetischen Energie der Photoelektronen bestimmt werden kann, ist charakteristisch für das Atom (genauer sogar für das Orbital), aus dem das Elektron stammt. Der zur Messung verwendete Analysator (meist ein Halbkugelanalysator) wird über elektrostatische Linsen und Gegenspannungen so eingestellt, dass ihn nur Elektronen einer bestimmten Energie passieren können. Für die XPS-Messung werden die Elektronen, die am Ende des Analysators noch ankommen, über einen Sekundärelektronenvervielfacher detektiert, so dass ein Spektrum entsteht, welches meistens in einem Graph durch die Auftragung der Intensität (Zählrate) über der kinetischen Energie der Photoelektronen dargestellt wird. Im nebenstehenden Bild ist ein typisches Spektrum gezeigt, bei dem die kinetische Energie der Elektronen über die lichtelektrische Gleichung bereits in die Bindungsenergie umgerechnet wurde. Die Nomenklatur der Beschriftung bedeutet hier: Fe2p für Elektronen, die aus einem Eisenrumpfniveau, genauer aus dem p-Orbital der L-Hauptschale stammen. Analog entspricht die gekürzte Schreibweise O1s den Elektronen aus dem s-Orbital der K-Hauptschale des Sauerstoffs. Das Spektrum lässt sehr schön die deutliche Spin-Bahn-Aufspaltung der beiden Fe2p-Emissionslinien Fe2p1/2 und Fe2p3/2 erkennen. Die zusätzlichen Bezeichnungen 3/2 bzw 1/2 entsprechen dabei dem Gesamtbahndrehimpuls des jeweiligen Elektrons in diesem Orbital. Eine vergleichbare Aufspaltung findet sich bei allen Elementen für die Emissionen aus p-, d- und f-Orbitalen. Quantitative Auswertung der MessungDie Intensität, also die Zählrate dieser Messungen, ist proportional zur Häufigkeit des Auftretens der verschiedenen Elemente in der Probe. Um nun die chemische Zusammensetzung eines Festkörpers zu bestimmen, muss man nur die Fläche unterhalb der beobachteten Linien, die charakteristisch für die Elemente sind, auswerten. Dabei sind allerdings einige messspezifische Besonderheiten zu beachten. So kann zum Beispiel ein Photoelektron, bevor es den Festkörper verlässt, weitere Elektronen anregen und dabei einen Teil seiner kinetischen Energie an diese abgeben. Diese sog. Sekundärelektronen besitzen praktisch keine diskrete Energieverteilung und tragen daher gleichmäßig zum Anwachsen des Untergrunds in einem XPS-Spektrum bei. Im Bild 3 kann man dieses Verhalten am stufenartigen Anwachsen (in Richtung größerer negativer Bindungsenergie) der Zählrate nach jeder Linie erkennen. Dieser Untergrund muss vor der Auswertung der Flächen über geeignete Methoden abgezogen werden, etwa durch Subtraktion eines linearen Untergrunds. Genauer ist der Untergrund-Abzug nach einer Methode, die auf D. A. Shirley[4] zurückgeht und Shirley-Untergrund-Korrektur genannt wird. Die genaueste (und aufwändigste) Methode besteht darin, den Verlauf des Untergrundes mittels der Elektronen-Energieverlustspektroskopie exakt zu bestimmen und das Ergebnis dieser Messung anschließend vom XPS-Spektrum zu subtrahieren; diese Methode wird als Tougaard-Untergrund-Korrektur bezeichnet[5]. Außerdem ist bei XPS-Messungen zu beachten, dass die Wahrscheinlichkeit für das Auslösen eines Photoelektrons energieabhängig, elementspezifisch und orbitalabhängig ist. Um dieser Tatsache Rechnung zu tragen, müssen die Werte für die Flächen, die unter den jeweiligen Linien ermittelt werden, um sog. Sensitivitätsfaktoren oder Wirkungsquerschnitte korrigiert werden, die in unterschiedlichen Tabellenwerken[6] zu finden sind. Weiterhin hängt die Wahrscheinlichkeit, dass von der Röntgenstrahlung erzeugte Photoelektronen den Festkörper tatsächlich verlassen und detektiert werden können, davon ab, wie häufig sie im Festkörper gestreut oder reabsorbiert werden. Diese Verlustrate hängt von der kinetischen Energie der Photoelektronen und der Zusammensetzung des Festkörpers ab. Man kann diesem Effekt durch die Berücksichtigung der mittleren freien Weglänge der Elektronen im Festkörper Rechnung tragen. Die entsprechenden Daten sind zumindest für die meisten Elemente und einfache Verbindungen tabelliert[7]. Unter Beachtung aller genannten Effekte ergibt die Auswertung des Spektrums in Bild 3 ein Eisen-zu-Sauerstoff-Verhältnis von 3:4, die Summenformel des untersuchten Materials ist also Fe3O4, es handelt sich um Magnetit. Erweiterte AnalyseEine weitere wesentliche Information über die chemischen Bindungsverhältnisse in der Probe beruht auf der Entdeckung von K. Siegbahn, dass die Spektren der Rumpfelektronen von der chemischen Umgebung des untersuchten Systems abhängen. In den XPS-Spektren desselben Elementes zeigen sich je nachdem, in welcher chemischen Form es vorliegt, Unterschiede in der Bindungsenergie eines Rumpfelektrons von bis zu einigen Elektronenvolt, dies wird als chemische Verschiebung (engl. chemical shift) bezeichnet. Beispielsweise kann so bestimmt werden, welchen Oxidationszustand ein Metallatom besitzt. Zusätzlich kann in vielen Fällen auch die Form der Spektren Aufschluss über den Valenzzustand eines Elementes geben. Die Photoemission begleiten weitere physikalische Prozesse, wie Photolumineszenz, oder auch das Auftreten von Auger-Elektronen uvm., die wiederum ihre Anwendung als eigene Messmethode gefunden haben. LichtquellenDie gängigsten Röntgenquellen, die in der XPS ihren Einsatz finden sind Al Kα- oder auch Mg Kα-Quellen, wobei exotischere Röntgenquellen allerdings auch Si-, Ti- oder Zr-Röntgenlinien erzeugen. In den letzten 20 Jahren hat sich die Verwendung von Synchrotronstrahlung, die sich aufgrund ihrer nahezu unbegrenzten Durchstimmbarkeit der Photonenenergie und Monochromie hervorragend als Anregungsquelle eignet, immer mehr durchgesetzt. Somit ist der Bereich der zugänglichen anregenden Photonenenergie von einigen wenigen diskreten Werten (z. B. Al Kα, hν = 1486,6 eV und Mg Kα, hν = 1253,6 eV) auf ein Kontinuum, das von einigen eV bis zu 20 keV reicht, erweitert worden. Technische DetailsDa es einen sehr hohen technischen (und damit finanziellen) Aufwand bedeutet, Messungen mit Hilfe von Synchrotronstrahlung durchzuführen, finden die zuvor erwähnten Röntgenröhren eine weite Verbreitung bei Standard-XPS-Analysen (siehe Bild 1). Die Energie der mit diesen Röntgenquellen erzeugten Photoelektronen liegt im Bereich zwischen 0 und 1500 eV, was für PES-Messungen bedeutet, dass die emittierten Elektronen aus einer maximalen Tiefe der untersuchten Probe, die zwischen 0 und 100 Å liegt, stammen. Der limitierende Faktor ist hier die mittlere freie Weglänge von Elektronen im Festkörper. Hier findet sich der Grund, warum die XPS vorwiegend zur Analyse von Festkörperoberflächen eingesetzt wird. Für solche Messungen ist in der Regel ein Basisdruck der Analysekammer im Bereich von Ultrahochvakuum (UHV) notwendig, der zwischen 0,5 und 5 · 10−10 hPa (Luftdruck auf Meereshöhe: ca. 1013 hPa) liegt. Diese Forderung nach extremen Vakuumbedingungen ergibt sich aus der unvermeidbaren Verunreinigung der Probe mit Adsorbaten aus der Umgebungsluft, wie z. B. Wasser oder auch Kohlenstoff, die mehrere µm betragen kann und somit keine Messung an der interessierenden Festkörperoberfläche erlauben würde. Um dieses Problem zu umgehen werden Proben in die UHV-Kammer verbracht und mit geeigneten Methoden, wie z. B. durch Sputtern, Tempern, Feilen oder auch Spalten von Einkristallen hochrein präpariert. Allerdings kann diese inhärente Schwäche der Messmethode zu einem starken Vorteil verwandelt werden. Die Informationstiefe der PES ist durch die mittlere freie Weglänge der Elektronen in einem Festkörper beschränkt. Für ein Metall z. B. beträgt diese gerade 1 bis 2 nm, was im Vergleich zur Eindringtiefe der Röntgenstrahlen (ca. 1–10 µm) um drei Größenordnungen kleiner ist. Es ist klar, dass nur die Photoelektronen vom Detektor erfasst werden können, die den Festkörper auch verlassen können. Der Intensitätsbeitrag zum Spektrum nimmt also mit zunehmender Tiefe exponentiell ab. Durch Veränderung des Winkels des Detektors gegenüber der zu messenden Probe kann eine extreme Oberflächensensitivität der Messung erreicht werden. Gerade im Bereich der Oberflächenpysik kann auf diese Art fast ausschließlich die erste Monolage einer Probe untersucht werden. ValenzbandspektroskopieDie Ultraviolettspektroskopie (engl. ultraviolet photoelectron spectroscopy, UPS) wird oft auch als Valenzbandspektroskopie bezeichnet, da die anregende Energie des Lichts (siehe theoretische Beschreibung) im UV-Bereich liegt und somit nur zum Auslösen von Elektronen des Valenzbandes fähig ist. Diese Energien sind natürlich der XPS-Messung ebenfalls zugänglich, nur kann durch eine geeignete Wahl der Lichtqelle (i. A. He-Gasentladungslampen) die kinetische Energie der so ausgelösten Photoelektronen mit extrem hoher Genauigkeit gemessen werden. UPS ist somit in der Lage, auch minimale Energieunterschiede von Molekülorbitalen oder auch der physikalischen Umgebung (z. B. Adsorption an Oberflächen) des spektroskopierten Moleküls aufzulösen. Bei der UPS steht also die Untersuchung der chemischen Struktur der Bindungen, die Mechanismen der Adsorption auf Substraten, oder auch die Bestimmung der Energie von Molekülvibrationen verschiedener Gase im Vordergrund. Winkelaufgelöste Messungen (ARPES)Mit Hilfe winkelaufgelöster Messungen, ARPES oder auch ARUPS (engl. angle-resolved UPS) genannt, wird nicht nur die Energie der Photoelektronen, sondern auch der Winkel unter dem sie die Probe verlassen gemessen. Auf diese Weise ist eine Bestimmung der Energie-Impulsbeziehung des Elektrons im Festkörper, also die Darstellung der Bandstruktur oder auch die Visualisierung von Fermiflächen, möglich. MessprinzipAlle bisher aufgeführten Methoden der PES detektieren die Photoelektronen unabhängig vom Winkel unter dem sie die Probe verlassen. Genau genommen wählt man bei diesen Messungen im Allgemeinen die Messposition des Analysators derart, dass vorwiegend Elektronen mit einem Austrittswinkel senkrecht zur Probenoberfläche detektiert werden können. Die Analysatoreinstellungen (genauer die Linsenspannungen der elektrostatischen Linsen, die Elektronenoptik) werde so eingestellt, dass sich ein sehr weiter Winkelakzeptanzbereich von ca. ±10° ergibt. Für die im Folgenden beschriebene Messmethode werden die Einstellungen des Analysators so verändert, dass Photoelektronen nur unter einem deutlich kleineren Winkel detektiert werden. Moderne Analysatoren erreichen dabei eine Winkelauflösung von weniger als 0,2° bei gleichzeitiger Energieauflösung von 1–2 meV. Ursprünglich wurden hohe Energie- und Winkelauflösung nur bei niedrigen Photoelektronenenergien im UV-Bereich erreicht, woraus sich dann auch der Name ARUPS ableitete. Gerade in den Jahren 1990–2000 wurde die Auflösung der PES-Analysatoren durch die Kombination von Mikrokanalplatten, phosphorisierender Platte und CCD-Kamera derart verbessert, dass auch bei weit höheren Photoelektronenenergien (Stand der Technik 2006: Ekin≈10keV) eine Bestimmung der Bandstruktur möglich wurde. Aufgrund der für den Analysator bedingten Forderung nach niedrigen Photonenenergien, die beispielsweise aus einer He-Lampe stammen, konnten ARUPS-Messungen nur die Bandstruktur der oberflächennahen Bereiche eines Festkörpers bestimmen. Zusammen mit der Verbesserung der Auflösung der Analysatoren und der Nutzung von hochenergetischem (Ekin > 500 eV), extrem monochromatischem Synchrotronlicht ist es seit ca. 2000 möglich die Bandstruktur des Volumens von kristallinen Festkörpern zu bestimmen. dies ist einer der Gründe, warum sich die ARPES in unserer Zeit zu einer der wichtigsten spektroskopischen Methoden zur Bestimmung der elektronischen Struktur von Festkörpern entwickelt hat. Qualitative Auswertung der MessungEine wesentliche Voraussetzung für die Gültigkeit der Aussage, dass ARPES die Bandstruktur eines kristallinen Festkörpers bestimmen kann, ist die Anwendbarkeit des Bloch-Theorems auf die beteiligten elektronischen Zustände, das heißt, dass sie sich durch einen Wellenzahlvektor k eindeutig charakterisieren lassen und die zugehörige Wellenfunktion die allgemeine Form: besitzt, wobei uk eine gitterperiodische Funktion ist. Diese Voraussetzung ist im Experiment nicht erfüllbar. Aufgrund des Übergangs von der Oberfläche der Probe zum Vakuum ist das System in senkrechter Richtung nicht translationsinvariant und damit der k Bei nicht zu starker Variation der Zustandsdichte (Bandstruktur) des untersuchten Kristalls senkrecht zur Oberfläche ist es also möglich die besetzte Zustandsdichte direkt zu messen. Um den Vergleich mit theoretischen Rechnungen zu erlauben, wird meistens in bestimmte Hochsymmetrierichtungen der Brillouin-Zone gemessen. Dazu wird der Kristall oft mittels LEED senkrecht zum Analysator orientiert, dann entlang einer der Richtungen gedreht und ein Energiespektrum der Photoelektronen aufgenommen. Mittels einer Mikrokanalplatte und einer CCD-Kamera können sogar gleichzeitig die Energie und der Winkel unter dem die Elektronen die Oberfläche verlassen, gemessen werden. Die Darstellung der Ergebnisse erfolgt meistens, indem man alle winkelabhängigen Spektren in einem Graphen darstellt, wobei die Energie und Intensität auf den Koordinatenachsen aufgetragen wird. Um die Winkelabhängigkeit verfolgen zu können werden die einzelnen Spektren in der Intensität verschoben, so dass die Dispersion beobachtbar wird. Eine alternative Darstellung ist eine Intensitätsverteilung mittels Farbkodierung, bei der Winkel und Energie auf den Koordinatenachsen und die Intensität als Farbabstufung verdeutlicht werden. Die nahezu vollständige Spektroskopie des Halbraumes über einer metallischen Probe nach der oben genannten Methode, erlaubt nun aus den Spektren die Fermi-Fläche des Elektronensystems des Kristalls zu bestimmen. Die Fermi-Fläche ergibt sich definitionsgemäß aus der Aneinanderfügung aller Punkte im Impulsraum, bei denen ein elektronisches Band die Fermi-Energie kreuzt (wie bei der Dispersionsrelation genügt es, sich bei der Definition der Fermi-Fläche auf die erste Brillouin-Zone zu beschränken). In PES-Messungen mit konstanter Photonenenergie entsprechen die Durchtrittspunkte im Allgemeinen Emissionsrichtungen, bei denen in den Spektren die Intensität an der Fermi-Energie besonders hoch ist. Daher reicht es häufig aus, die Intensitätsverteilung bei EF in Abhängigkeit vom Emissionswinkel Θ zu bestimmen, ohne dabei den genauen Bandverlauf berücksichtigen zu müssen. Photoelektronenbeugung (XPD)
Die Photoelektronenbeugung oft mit PED, PhD oder auch XPD (engl. x-ray photoelectron diffraction) abgekürzt, ist eine Methode, um die Struktur von kristallinen Oberflächen oder die räumliche Lage von Adsorbaten auf Oberflächen zu bestimmen. MessprinzipGrundlage des Messverfahrens ist wieder die Photoelektronenspektroskopie, wobei die Intensität der Photoelektronen abhängig vom Emissionswinkel bestimmt wird. Allerdings ist hier nicht wie bei der winkelabhängigen PES der Fokus auf dem Impuls des Photoelektrons, sondern die Interferenz der Wellenfunktion des Photoelektrons. In Abhängigkeit von Emissionsrichtung und kinetischer Energie des Photoelektrons findet man Intensitätsunterschiede, Modulationen genannt. Diese Intensitätsmodulationen entstehen durch konstruktive und destruktive Interferenz zwischen der Elektronenwelle, die den Detektor auf direktem Weg erreicht (Referenzwelle), und solchen, die aus ein- oder mehrfach an der Umgebung des emittierenden Atoms elastisch gestreuten Wellen (Objektwellen) auftreten. Die Gangunterschiede und Intensitäten der einzelnen Wellen hängen von der geometrischen Anordnung und der Art der Nachbaratome ab. Bei einer ausreichenden Anzahl von gemessenen Intensitäten lässt sich aus den Modulationen die geometrische Struktur bestimmen, indem die experimentell gemessenen Modulationen mit entsprechenden Simulationen verglichen werden. Bei inelastisch gestreuten Wellen fehlt die feste Phasenbeziehung zur Referenzwelle, deswegen tragen sie nicht zur Interferenz bei. Da die Streuung der Elektronen vor Allem bei hohen Energien in Vorwärtsrichtung am stärksten ist, kann man im einfachsten Fall die Streuung der Elektronen an den Atomen unterhalb des ionisierten Atoms vernachlässigen. Bei der Streuung an den Atomen weiter oben werden die Photoelektronen in Richtung dieser Atome fokussiert („Vorwärts-Fokussierung“). AnwendungenDie einfachsten Anwendungen beruhen auf der Vorwärts-Fokussierung durch Atome oberhalb des photoionisierten Atoms. Damit lässt sich bestimmen, ob bestimmte Atome unmittelbar an der Oberfläche oder tiefer sitzen, und bei adsorbierten Molekülen, ob oberhalb einer Atomsorte noch andere Atome (und in welcher Richtung) sitzen. Mittels der XPD kann die kristallographische Struktur von Metall- und Halbleiteroberflächen bestimmt werden. Außerdem erhält man Informationen über die räumliche Lage von Molekülen auf Oberflächen, der Bindungslängen und Bindungswinkel. Vergleich mit komplementären TechnikenIm Vergleich zu anderen Techniken zur Bestimmung von Strukturen an Oberflächen ist Photoelektronenbeugung
Photoelektronenmikroskopie (PEEM)Eine weitere verbreitete Anwendung der PES ist die Photoelektronenmikroskopie kurz PEEM (engl. photo emission electron microscopy) genannt. Hier werden durch den Photoeffekt Elektronen aus der Probe herausgelöst, allerdings wird bei der Detektion nicht die Anzahl der Elektronen einer durch den Analysator ausgewählten kinetischen Energie gemessen, sondern man interessiert sich vielmehr für die Intensitätsverteilung der Photoelektronen eines Bereiches der Probe. MessprinzipDas PEEM sammelt die emittierten Photoelektronen mittels eines starken elektrostatischen Feldes zwischen Probe und Abbildungssäule, wobei das Elektronenbild im Anschluss mit mehreren koaxialen Linsen vergrößert wird. Die so ausgewählten Elektronen treffen nun auf einen Leuchtschirm und erzeugen ein Bild, das wiederum über eine CCD-Kamera abgebildet werden kann. Der direktere Weg der Abbildung geht über eine Mikrokanalplatte (große Anzahl von zweidimensional angeordneten Kanalelektronenvervielfacher (engl. channeltron)). Als Lichtquellen werden Quecksilber- (hν = 4,9 eV) und Deuteriumlampen (hν = 6,4 eV) genutzt. Auswertung der MessungDie Variation der Intensität der Elektronenausbeute wird bei der Messung direkt in Echtzeit am Bildschirm dargestellt. Sie beruht im allgemeinen beim PEEM auf den unterschiedlichen Austrittsarbeiten der untersuchten topographischen Bereiche der Probe. Auf diese Art können verschiedene Eigenschaften (z. B. Topografie der Oberfläche, Unterschiede der Austrittsarbeit verschiedener Stoffe, chemische Zusammensetzung und magnetische Feldstärke) durch eine Kontrastabstufung in Echtzeit mit einer Auflösung von bis zu 20 nm sichtbar gemacht werden. XPS-Mikroskop oder auch μ-ESCADurch den Einbau eines Mikroanalysators in den Strahlengang, der die kinetische Energie der Photoelektronen selektiert (analog zur normalen PES), sowie durch die Verwendung von schmalbandigen und kurzwelligen Anregungslichtquellen, wie z. B. Synchrotronstrahlung, ist es möglich, auch lateral aufgelöste XPS durchzuführen. Die Bezeichnung μ-ESCA beschreibt die chemische Analyse eines μm-großen Bereiches der Probe. Damit ist sowohl eine Bestimmung der Elementzusammensetzung der Probe als auch die Untersuchung lokaler Unterschiede der elektronischen Eigenschaften möglich. Spinaufgelöste PhotoelektronenspektroskopieMessungen in Resonanz (ResPES)Grundsätzlich ist der Verlauf des Photoemissionsspektrums, insbesondere das des Valenzbandes, abhängig von der zur Anregung verwendeten Photonenenergie. Überstreicht man mit der Photonenenergie den Bereich einer Röntgenabsorptionskante, so sind die Änderungen im Allgemeinen besonders stark ausgeprägt. Grund dafür sind Resonanzeffekte, die durch die Wechselwirkung zweier oder mehrerer verschiedener Endzustände, genauer von Kontinuumszuständen mit diskreten Niveaus, entstehen und so den Gesamtphotoemissionswirkungsquerschnitt beeinflussen. Trägt man die Photoemissionsintensität einer ausgewählten spektralen Struktur gegen die Photonenenergie auf, so erhält man im Allgemeinen asymmetrische Anregungsprofile, so genannte Fano-Resonanzen. Form und Intensität dieser Profile können Aufschluss über den elementaren Charakter der spektralen Struktur, über Details der chemischen Bindung und über die Wechselwirkungen der beteiligten Zustände geben. Inverse Photoelektronenspektroskopie (IPES)Im Gegensatz zur PES werden bei der inversen Photoelektronenspektroskopie Elektronen bekannter Energie auf die Probe beschleunigt und die dabei emittierten Photonen als Bremsstrahlung detektiert. Hieraus lässt sich die unbesetzte Zustandsdichte und die Bandstruktur oberhalb des chemischen Potentials (oberhalb der Fermi-Energie) ermitteln. Analog zu ARUPS erhält man bei IPES im UV-Bereich die k-Information aus der Einfallsrichtung der anregenden Elektronen. Apparativ setzt sich ein IPES-Spektrometer aus einer einfachen Elektronenkanone und einem Photonendetektor mit Bandpassfilter oder Monochromator zusammen. Zumeist wird bei Labormessungen die kinetische Energie (Primärenergie) der Elektronen variiert und die Photonenenergie bei der Detektion konstant gehalten. Man spricht in diesem Fall vom Isochromaten-Modus, wovon sich auch die Bezeichnung BIS, Bremsstrahlungsisochromaten-Spektroskopie, ableitet. Den häufigsten Einsatz finden für Energien im UV-Bereich Bandpassfilter vom Geiger-Müller-Typ, bei dem ein Erdalkalifluoridfenster als Tiefpass (z. B. CaF2 oder SrF2) und ein geeignetes Zählgas (z. B. I2 oder CS2) als Hochpass kombiniert werden. Detektionsenergie und Bandpassbreite ergeben sich aus der Transmissionsschwelle des Fenstermaterials bzw. aus der molekularen Photoionisationsschranke des Zählgases (etwa 9,5 eV). Die Bandpassbreite bestimmt im Wesentlichen die Spektrometerauflösung. Andere Detektortypen kombinieren das Erdalkalifluoridfenster mit einem geeignet beschichten Kanalelektronenvervielfacher (z. B. mit NaCl oder KBr). Wegen des geringen Wirkungsquerschnittes des inversen Photoemissionsprozesses ist die typische Zählrate im Vergleich zur Photoelektronenspektroskopie sehr klein. Daher lassen sich bei der IPES auch keine vergleichbaren Energieauflösungen erreichen, da das Signal mit der Bandpassbreite linear abnimmt. Typische Werte für die Auflösung liegen bei wenigen Hundert Millielektronenvolt, sind also zwei Größenordnungen schlechter als bei UPS. Detektoren mit Gittermonochromatoren erreichen prinzipiell deutlich bessere Werte und sind wegen ihrer durchstimmbaren Photonenenergie wesentlich flexibler einsetzbar, sind aber sehr viel teurer und größer als die anderen Detektortypen. Siehe auchFußnoten und Einzelnachweise
Literatur
Kategorien: Oberflächenphysik | Spektroskopie |
| Dieser Artikel basiert auf dem Artikel Photoelektronenspektroskopie aus der freien Enzyklopädie Wikipedia und steht unter der GNU-Lizenz für freie Dokumentation. In der Wikipedia ist eine Liste der Autoren verfügbar. |




 -Anteil des gemessenen Wellenzahlvektors keine gute
-Anteil des gemessenen Wellenzahlvektors keine gute 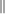 -Anteil erhalten, da sowohl das Kristallpotenzial als auch das Vakuum parallel zur Oberfläche gitterperiodisch bleiben. Folglich kann man den Betrag des Wellenzahlvektors in diese Richtung direkt angeben:
-Anteil erhalten, da sowohl das Kristallpotenzial als auch das Vakuum parallel zur Oberfläche gitterperiodisch bleiben. Folglich kann man den Betrag des Wellenzahlvektors in diese Richtung direkt angeben: