Maschinelles Lernens beschleunigt die Bewertung von Katalysatoren von Monaten auf Millisekunden
Modell zur Identifizierung kostengünstiger Katalysatoren, die Biomasse in Kraftstoffe und nützliche Chemikalien umwandeln
Anzeigen


Wenn Sie das nächste Mal auf einer Landstraße an Bauernhöfen oder Wiesen und Teichen vorbeifahren, schauen Sie sich um. Sie sind eine reiche Quelle für Biomasse. Dazu gehören Mais, Sojabohnen, Zuckerrohr, Rutenhirse, Algen und anderes Pflanzenmaterial. Diese kohlenstoffreichen Materialien können in flüssige Brennstoffe und Chemikalien mit vielen Anwendungsmöglichkeiten umgewandelt werden. In den Vereinigten Staaten gibt es beispielsweise genug Biomasse, um erneuerbaren Düsenkraftstoff für den gesamten Flugverkehr herzustellen.

Von Monaten zu Millisekunden: Aus langsam wird schnell (Symbolbild)
Computer-generated image
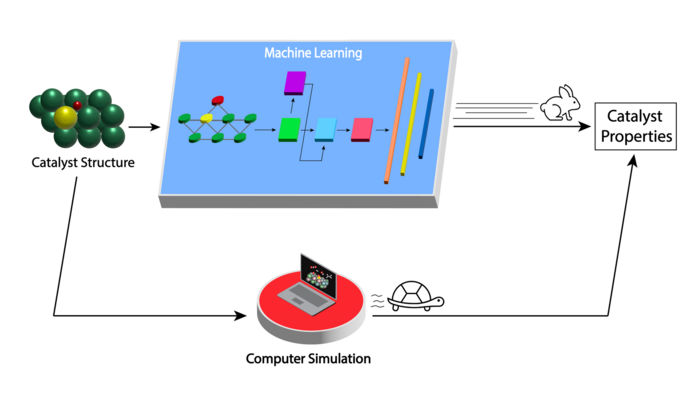
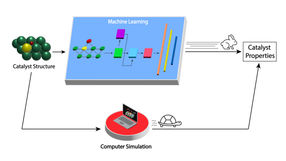
Das neu entwickelte maschinelle Lernmodell beschleunigt die Bewertung der Eigenschaften von Molybdäncarbid-Katalysatoren für die Umwandlung von Biomasse in nützliche Produkte (oberer Pfad) im Vergleich zu den derzeitigen Computersimulationsmethoden (unterer Pfad) erheblich.
Image by Argonne National Laboratory

Ein großer Stolperstein ist derzeit der Mangel an wirksamen, kostengünstigen Katalysatoren, die für die Umwandlung von Biomasse in Biokraftstoff oder andere nützliche Produkte benötigt werden. Forscher des Argonne National Laboratory des US-Energieministeriums (DOE) berichten, dass sie ein auf künstlicher Intelligenz basierendes Modell entwickelt haben, um den Prozess der Entwicklung eines kostengünstigen Katalysators auf der Grundlage von Molybdänkarbid zu beschleunigen.
"Biomasse ist ein organisches Material, das heißt, es ist voller Kohlenstoff", sagte Rajeev Assary, Gruppenleiter in der Materials Science Division (MSD) von Argonne. Das ultimative Ziel ist es, diesen Kohlenstoff kostengünstig in nützliche Produkte für die Gesellschaft umzuwandeln, in diesem Fall in Biokraftstoff und Chemikalien wie biologisch abbaubaren Kunststoff. Diese Produkte machen den Einsatz fossiler Brennstoffe überflüssig."
Derzeit können Wissenschaftler durch die Behandlung von Rohbiomasse bei hohen Temperaturen ein erdölähnliches Produkt namens Pyrolyseöl herstellen. Das dabei entstehende Produkt weist jedoch einen sehr hohen Sauerstoffgehalt auf. Dieser Sauerstoff ist unerwünscht und wird daher durch eine Reaktion entfernt, die durch den Einsatz eines Molybdänkarbid-Katalysators ermöglicht wird. Ein großes Problem besteht jedoch darin, dass die Oberfläche dieses Katalysators Sauerstoffatome absorbiert, die sich an der Oberfläche anlagern und die Leistung des Katalysators beeinträchtigen.
Eine vorgeschlagene Lösung besteht darin, dem Molybdänkarbid eine kleine Menge eines neuen Elements wie Nickel oder Zink beizumischen. Dieses Dotierungselement schwächt die Bindung der Sauerstoffatome auf der Katalysatoroberfläche und verhindert so eine Vergiftung des Katalysators.
"Das Problem besteht darin, die richtige Kombination aus Dotierstoff und Oberflächenstruktur zu finden", sagt Hieu Doan, wissenschaftlicher Mitarbeiter bei MSD. Molybdänkarbid hat eine sehr komplizierte Struktur. Wir haben daher Supercomputing mit theoretischen Berechnungen kombiniert, um das Verhalten nicht nur der Oberflächenatome zu simulieren, die sich mit Sauerstoff verbinden, sondern auch der Atome in der Nähe."
Mit Hilfe von Simulationen, die auf dem Theta-Supercomputer von Argonne durchgeführt wurden, erstellte das Team eine Datenbank mit 20.000 Strukturen für die Bindungsenergien von Sauerstoff an dotiertem Molybdäncarbid. Bei den Simulationen wurden mehrere Dutzend Dotierungselemente und über hundert mögliche Positionen für jedes Dotierungselement auf der Katalysatoroberfläche berücksichtigt. Theta ist Teil der Argonne Leadership Computing Facility, einer Nutzereinrichtung des DOE Office of Science.
Mit dieser Datenbank trainierten sie dann ein Deep-Learning-Modell. Deep Learning ist eine Form des maschinellen Lernens, bei der der Computer lernt, Probleme zu lösen, indem er zunächst eine große Menge von Daten analysiert. Mit unserem Deep-Learning-Modell können wir nun präzise und kostengünstige Berechnungen für Zehntausende von Strukturen in Millisekunden durchführen, während wir mit herkömmlichen Berechnungsmethoden nur einige tausend Katalysatorstrukturen über Monate hinweg bewerten können", so Doan. "Das ist Material-Screening auf Steroiden."
Das Team hat die Ergebnisse seiner Simulationen auf atomarer Ebene und seines Deep-Learning-Modells an das Chemical Catalysis for Bioenergy Consortium geschickt. Sie werden Experimente durchführen, um eine kleine Anzahl von Katalysatorkandidaten zu bewerten.
"Wir hoffen, dass wir in naher Zukunft mehr als eine Million Strukturen und verschiedene Bindungsatome, wie z. B. Wasserstoff, verarbeiten können", so Assary. "Wir wollen denselben Berechnungsansatz auch auf Katalysatoren für andere Dekarbonisierungstechnologien anwenden, etwa für die Umwandlung von Wasser in sauberen Wasserstoff als Kraftstoff."
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft