Physiker simulieren das Kristallwachstum im Computer
Anzeigen



Das Wachstum von Halbleiterkristallen wird am Lehrstuhl für Theoretische Physik III (Computational Physics) der Universität Würzburg mit Hilfe von Computersimulationen untersucht. Ein Schwerpunkt liegt auf Materialien, die aus verschiedenen Atomsorten bestehen, wie es zum Beispiel bei den modernen Verbindungshalbleitern der Fall ist.
Für die Entwicklung neuartiger elektronischer Bauelemente - zum Beispiel von verbesserten Laserdioden für CD-Spieler - benötigt man perfekte Kristalle aus Halbleitermaterialien. Eine wichtige Technik für die Herstellung solcher Kristalle im Labor ist die Molekularstrahlepitaxie: Dabei werden in einer Vakuumkammer geringe Mengen des gewünschten Materials in einem Ofen verdampft. Die Atome treffen auf die Oberfläche einer Schicht, in die sie schließlich eingebaut werden und die dadurch wächst.
"Ein fundiertes theoretisches Verständnis der Wachstumsprozesse sollte es ermöglichen, diese experimentellen Techniken systematisch zu verbessern", sagt der Würzburger Physiker Dr. Michael Biehl. In diesem Zusammenhang stelle die Simulation im Computer ein wichtiges Werkzeug dar.
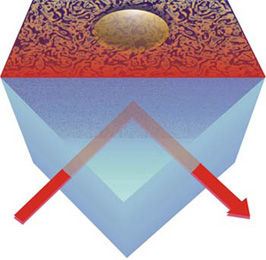
Bei der Molekularstrahlepitaxie sollen in der Regel möglichst glatte Schichten des gewünschten Materials entstehen. In der Praxis zeigen sich aber häufig Abweichungen von diesem Ideal: Es bilden sich unregelmäßige, raue Oberflächen oder sogar kleine Hügel, die an Pyramiden oder Kegel erinnern.
"In der Simulation kann man nun - anders als beim Experiment - ganz bestimmte Prozesse verbieten oder bevorzugen", so Dr. Biehl. Auf diese Weise lasse sich zum Beispiel herausfinden, welche Rolle es für die Bildung der Hügel spielt, wenn Atome an den Kanten des wachsenden Kristalls gewissermaßen herunterklettern.
In diesem Zusammenhang stellen sich weitere Fragen: Welchen Einfluss hat die Temperatur oder die Wachstumsgeschwindigkeit auf die Hügelbildung? Unter welchen Bedingungen entstehen möglichst glatte Flächen? Solchen und ähnlichen Fragen gehen Dr. Biehl, Prof. Dr. Wolfgang Kinzel und Diplom-Physiker Martin Ahr zusammen mit anderen Kollegen im Rahmen eines Projektes nach, das von der Deutschen Forschungsgemeinschaft gefördert wird.
Es sollen Modelle weiterentwickelt werden, welche die Simulation relativ großer Systeme mit vertretbarem Zeitaufwand erlauben. Denn bislang stoßen die Wissenschaftler noch rasch an die Leistungsgrenzen der heutigen Rechner. Schließlich müssen möglichst große Systeme aus sehr vielen Atomen simuliert werden, um die interessanten Effekte überhaupt beobachten zu können. Mit der Zahl der Teilchen wächst natürlich auch die benötigte Rechenzeit.
Deshalb ist es von besonderer Bedeutung, effiziente Modelle und schnelle Computerprogramme zu entwickeln. Der wichtigste und zugleich schwierigste Schritt besteht darin, geeignete Modellvorstellungen zu erarbeiten: Sie sollen einerseits die komplizierten physikalischen Prozesse soweit vereinfachen, dass das Problem lösbar wird. Andererseits müssen sie natürlich immer noch die wichtigsten Materialeigenschaften wiedergeben.
Dr. Biehl: "Bei einer sehr erfolgreichen Klasse von Modellen werden die simulierten Teilchen nur auf den Plätzen eines fest vorgegebenen Kristallgitters bewegt. Nach bestimmten Spielregeln werden Atome wie Bauklötzchen auf das Gitter gesetzt, können dort herumwandern, sich an andere Teilchen anlagern und zur Ruhe kommen." Die Bewegungsregeln ergeben sich dabei aus den physikalischen Wechselwirkungen der Teilchen, die stark vereinfacht durch anziehende oder abstoßende Kräfte repräsentiert werden.



Weitere News aus dem Ressort Wissenschaft























































