Neue Methode verbessert die Genauigkeit von maschinell gelernten Potenzialen zur Katalysator-Simulation
Brückenschlag zwischen Genauigkeit und Effizienz bei der Katalysatorentwicklung
Anzeigen



Katalysatoren spielen in der modernen Fertigung eine unverzichtbare Rolle. Mehr als 80 % aller hergestellten Produkte, von Arzneimitteln bis zu Kunststoffen, sind in irgendeinem Stadium der Produktion auf katalytische Prozesse angewiesen. Vor allem Übergangsmetalle zeichnen sich als hochwirksame Katalysatoren aus, da sie aufgrund ihrer teilweise gefüllten d-Orbitale leicht Elektronen mit anderen Molekülen austauschen können. Genau diese Eigenschaft macht es jedoch schwierig, sie genau zu modellieren, da eine präzise Beschreibung ihrer elektronischen Struktur erforderlich ist.
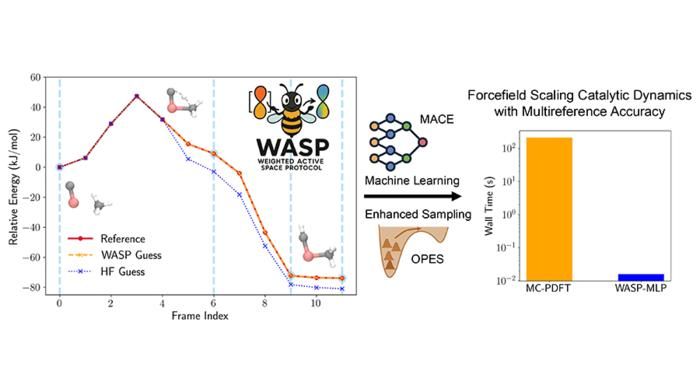
Genauigkeit und Beschleunigung mit dem Weighted Active Space Protocol (WASP) für die Methanaktivierung auf Titankarbid.
Figure courtesy of Seal et al.
Die Entwicklung effizienter Übergangsmetallkatalysatoren, die unter realistischen Bedingungen funktionieren, erfordert mehr als eine statische Momentaufnahme einer Reaktion. Stattdessen müssen wir das dynamische Bild erfassen, wie sich Moleküle bei unterschiedlichen Temperaturen und Drücken bewegen und interagieren, wobei die atomare Bewegung die katalytische Leistung grundlegend beeinflusst.
Um diese Herausforderung zu meistern, hat das Labor von Prof. Laura Gagliardi an der University of Chicago Pritzker School of Molecular Engineering (UChicago PME) und dem Fachbereich Chemie ein leistungsfähiges neues Tool entwickelt, das Theorien der elektronischen Struktur und maschinelles Lernen nutzt, um die katalytische Dynamik von Übergangsmetallen sowohl genau als auch schnell zu simulieren.
"In den letzten zehn Jahren haben maschinell erlernte Potenziale die Art und Weise, wie wir die Molekulardynamik simulieren, erheblich verbessert und bieten Geschwindigkeit und Skalierbarkeit. Die genaue Erfassung der elektronischen Struktur von Übergangsmetallkatalysatoren ist jedoch nach wie vor eine ungelöste Herausforderung. Unsere neue Methode überbrückt diese Lücke, indem sie Methoden der Multireferenz-Quantenchemie mit maschinell erlernten Potenzialen kombiniert und so sowohl Genauigkeit als auch Effizienz bietet". sagte Gagliardi.
Die Ergebnisse wurden in den Proceedings of the National Academy of Sciences veröffentlicht.
Maschinelles Lernen zur Beschleunigung von Simulationen
In den letzten zehn Jahren hat die Gruppe von Gagliardi die Multikonfigurations-Paar-Dichtefunktionaltheorie (MC-PDFT) entwickelt, eine quantenchemische Methode, mit der sich die komplizierten elektronischen Strukturen von Übergangsmetallreaktionen beschreiben lassen. Die MC-PDFT bietet zwar eine hohe Genauigkeit, ist aber für die Simulation der Dynamik katalytischer Systeme unerschwinglich langsam - ein entscheidender Schritt bei der Vorhersage, wie sich Katalysatoren unter realistischen Bedingungen tatsächlich verhalten.
Um diese Herausforderung zu meistern, wandte sich das Team maschinell erlernten interatomaren Potenzialen (ML-Potenzialen) zu, die die molekulare Dynamik mit bemerkenswerter Effizienz erfassen können. ML-Potenziale sind in der Materialwissenschaft weit verbreitet, aber bis jetzt wurden sie noch nie erfolgreich mit Multireferenzmethoden wie MC-PDFT kombiniert.
Der Grund dafür liegt in einem seit langem bestehenden Hindernis: der Konsistenz der Beschriftung. Modelle des maschinellen Lernens benötigen für jede Molekülgeometrie entlang eines Reaktionsweges eindeutige und zuverlässige Eigenschaftskennzeichnungen, wie z. B. aus Wellenfunktionen abgeleitete Energien und Kräfte. Für quantenchemische Methoden mit mehreren Referenzen war die eindeutige Zuweisung solcher Bezeichnungen bisher ein ungelöstes Problem.
Um diese Herausforderung zu meistern, entwickelte der Doktorand Aniruddha Seal unter der gemeinsamen Beratung von Gagliardi und Prof. Andrew Ferguson einen neuartigen Algorithmus, der konsistente Wellenfunktionen für neue Geometrien als gewichtete Kombination von Wellenfunktionen aus zuvor untersuchten Molekülstrukturen erzeugt. Je näher eine neue Geometrie an einer bekannten Geometrie liegt, desto stärker ähnelt ihre Wellenfunktion derjenigen der bekannten Struktur. Dieser Ansatz stellt sicher, dass jedem Punkt entlang eines Reaktionsweges eine eindeutige, konsistente Wellenfunktion zugewiesen wird, so dass ML-Potenziale genau auf Multireferenzdaten trainiert werden können.
"Stellen Sie sich das vor wie das Mischen von Farben auf einer Palette", erklärt Seal. "Wenn ich einen Grünton erzeugen will, der eher blau ist, nehme ich mehr blaue Farbe und nur ein wenig Gelb. Wenn ich einen Farbton möchte, der eher gelb ist, kehrt sich das Verhältnis um. Je näher meine Zielfarbe an einer der Grundfarben liegt, desto stärker beeinflusst sie die Mischung. WASP funktioniert auf die gleiche Weise: Es kombiniert Informationen aus nahe gelegenen Molekularstrukturen und gewichtet die ähnlichsten stärker, um eine genaue Vorhersage für die neue Geometrie zu erstellen."
Diese Innovation bildet die Grundlage für das Weighted Active Space Protocol (WASP), ein Verfahren, das die Genauigkeit der MC-PDFT mit der Effizienz des maschinellen Lernens verbindet. Es wurde in enger Zusammenarbeit mit der Parrinello-Gruppe am Italian Institute of Technology in Genua entwickelt und vereint Fachwissen auf dem Gebiet der elektronischen Strukturtheorie und des maschinellen Lernens. WASP ermöglicht dramatische Geschwindigkeitssteigerungen: Simulationen mit Multireferenzgenauigkeit, die früher Monate dauerten, können jetzt in wenigen Minuten abgeschlossen werden.
Auswirkungen: Brückenschlag zwischen Genauigkeit und Effizienz beim Katalysatordesign
Durch die Verbindung von Genauigkeit und Geschwindigkeit öffnet WASP die Tür zur Entwicklung von Katalysatoren, die realistischen Bedingungen standhalten können - hohen Temperaturen und hohem Druck. Übergangsmetalle sind für zahllose großtechnische Prozesse von zentraler Bedeutung, aber ihre Komplexität hat das rationale Design von Katalysatoren zu einer Herausforderung gemacht.
Ein Paradebeispiel ist das Haber-Bosch-Verfahren, bei dem Eisen als Katalysator für die Umwandlung von Stickstoff und Wasserstoff in Ammoniak dient. Obwohl dieser Eisenkatalysator vor mehr als einem Jahrhundert entwickelt wurde, dominiert er noch immer die weltweite Ammoniakproduktion. Mit WASP haben die Forscher nun die Möglichkeit, Alternativen zu erforschen, die die Effizienz steigern, Nebenprodukte reduzieren und die Umweltkosten senken könnten.
Bisher wurde WASP erfolgreich für die thermisch aktivierte Katalyse eingesetzt - also für Reaktionen, die durch Wärme angetrieben werden. Das nächste Ziel ist die Anpassung der Methode an lichtaktivierte Reaktionen, die für die Entwicklung neuer Photokatalysatoren unerlässlich sind. Photokatalysatoren bieten ein enormes Potenzial für Technologien, von der Wasseraufbereitung bis zur Energieerzeugung.
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung




Weitere News aus dem Ressort Wissenschaft


























































